Microsoft word - lidia brunetto's phd thesis.doc


Università degli Studi di Catania
Scuola Superiore di Catania
International PhD
Investigation of Cancer Stem Cells (CSCs)-derived
exosomes and their influence on the
tumor microenvironment
Coordinator of PhD Tutor
Prof. Daniele Condorelli
Prof. Ruggero De Maria
a.a. 2008/2011
Agli uomini della mia vita
Valerio e Riccardo
1. Tumor Microenvironment
2. Cancer Stem Cells (CSCs) and Lung
3. CD133 stem cell marker
4. CSC-derived Exosomes
Working Hypothesis
Materials and Methods
1. Lung Cancer Stem lines
2. Adherent Cell lines
3. Virus production and Infection
4. Isolation of Lung Cancer Stem Cells (LCSCs)-derived exosomes
5. Western Blot analysis
6. FACS analysis
7. Immunoprecipitation (IP)
8. Screening for optimal cellular system to investigate the uptake of CD133
containing particles 36
9. Exosomes uptake assay
10. Immunofluorescence (IF)
11. Transfection procedures
12. QReal Time RT-PCR
1. Characterization of Lung cancer stem cells (LCSCs)
2. Biochemical characterization of exosomes
3. CD133 is present on the surface of exosomes
4. CD133 is transported through exosomes into HEK293T cells
5. Uptake Kinetic of exosomes by HEK293T cells
6. CD133 can activate intracellular parhways
7. Tetracycline-dependent regulatory system
8. CD133 mediates the transfer of biological materials from cell to cell
9. Secretory exosomes are active vehicles for intercellular miRNA transfer
10. Immunoprecipitated CD133 positive exosomes contain miR-146a
Discussion
References
Abstract
Exosomes are membrane nanovesicles of endocytic origin that can be released to the
extracellular environment by many different cells, included the tumor cells.
Exosomes have been suggested to have a number of different functions and are
believed to take part in the communication between cells. The aim of this thesis was
to assess the composition and functions of Lung cancer stem cells (LCSCs)-derived
exosomes, with focus on the content of typical proteins, microRNAs and cell-to-cell
communication. We were able to set up an isolation protocol to obtain the exosomes
from LCSCs supernatant. The exosomes have a distinct composition of surface
proteins. By western blot analysis (WB) we characterized LCSCs-derived exosomes
for the presence of typical exosomes markers. We found that, in addition to well
known markers, such as Tsg101 and CD81, LCSC-derived exosomes are enriched in
CD133 protein. Immunoprecipitation (IP) experiments revealed that by
immunoprecipitating the culture SN of CD133 expressing cells with an antibody
against CD133 protein, typical exosomal markers, such as Tsg101 and Rab5b, are
contained in the precipitate. This suggests that CD133 is contained in bona fide
exosomal particles and that these proteins might have a physiological role in the
biological function of exosomes. The structure of CD133 protein shows homologies
with proteins involved in plasma membrane fusion, suggesting, that CD133 might
play an active role in the transport of biological information from one to another cell.
Immunofluorescence (IF) experiments and FACS analysis revealed that exosomal
vesicles, contained in the supernatant, are able to transfer CD133 protein from
expressing to non-expressing cells. Therefore we established an experimental system
in order to study a transport of proteins or genetic from cell to cell. Results of co-
culture experiments, using a tetracycline-dependent regulatory system, showed an
interchange of information between stably transduced cell lines. We wanted also
investigate if CD133 enriched SN is able to alter the phosphorylation status of
recipient cells. We found that the recipient cells, after incubation with CD133
enriched SN, show a marked increase in MAP kinase pathways.
In addition, transfer experiments suggest, that miR146a contained in LCSC136-
derived exosomes is shuttled to HEK293T recipient cells. Furthermore the transfer of
miR146a is increased by CD133 overexpression in LCSC136 cells.
In order to verify if CD133 containing exosomes, indeed transport microRNAs, we
performed immunoprecipitation experiments of CD133 enriched SN, using CD133
antibody. We found, that the CD133 positive membrane fraction isolated by
immunoprecipitation also contained miR146a. This result revealed that we didn't
isolate just protein aggregates, but CD133 positive exosomes, which are able to
transport microRNAs.
In summary, CSC-derived exosomes are bioactive molecules mediating the
interchange of informations and are able to stimulate target cells in order to develop
tumor capacities. This function can be enhanced by the presence of CD133, which
positively regulates the transport of bioactive molecules such as microRNAs between
cells. These new findings suggest a new, pro-tumorigenic role to this well known
cancer stem cell marker.
1. Tumor Microenvironment
In order to understand tumor biology in more detail, it is essential to further
investigate the complex molecular networks and cross talk between different
components of the tumor microenvironment and the tumor cells themselves. For a
long time cancer has been considered as a disease consisting of transformed cells,
which have acquired cell autonomous hyperproliferative and invasive capacities.
Emerging evidence indicates that we need to consider carcinogenesis and tumor
progression not as cell autonomous, cancer cell-centered condition, but rather as a
disease involving complex heterotypic multicellular interactions within a newly
formed tissue, the cancer tissue. Hence, the concept of tumor microenvironment as
an integrated and essential part of the cancer tissue was coined (1. Bissell et al.,
2002; 2. Hanahan and Weinberg, 2000).
The tumor microenvironment contains a number of distinct cell types (Figure 1),
including endothelial cells and their precursors, pericytes, smooth muscle cells,
fibroblasts, myofibroblasts, neutrophils, eosinophils, basophils, mast cells, T and B
lymphocytes, natural killer cells and antigen presenting cells (APC) such as
macrophages and dendritic cells (3. Coussens and Werb, 2002).
Experimental data demonstrated a role for these individual components, in particular
endothelial cells, macrophages, and cancer-associated fibroblasts, in promoting
tumor growth and progression. While most cellular components of the immune
system are in principle capable of rejecting tumors, they are enslaved by cancer cells
to promote their growth and invasion. Importantly, the stromal mass forms about half
of most malignant solid tumors. Fibroblasts contributing to the tumor stroma have
been termed peritumoral fibroblasts, reactive stroma, cancer-associated fibroblasts
(CAF) and myofibroblasts (4. De Wever et al., 2008).
Fibroblastic cells adjacent to cancer cell nests, generally express a-smooth muscle
actin (a-SMA), an important marker for differentiated myofibroblasts.
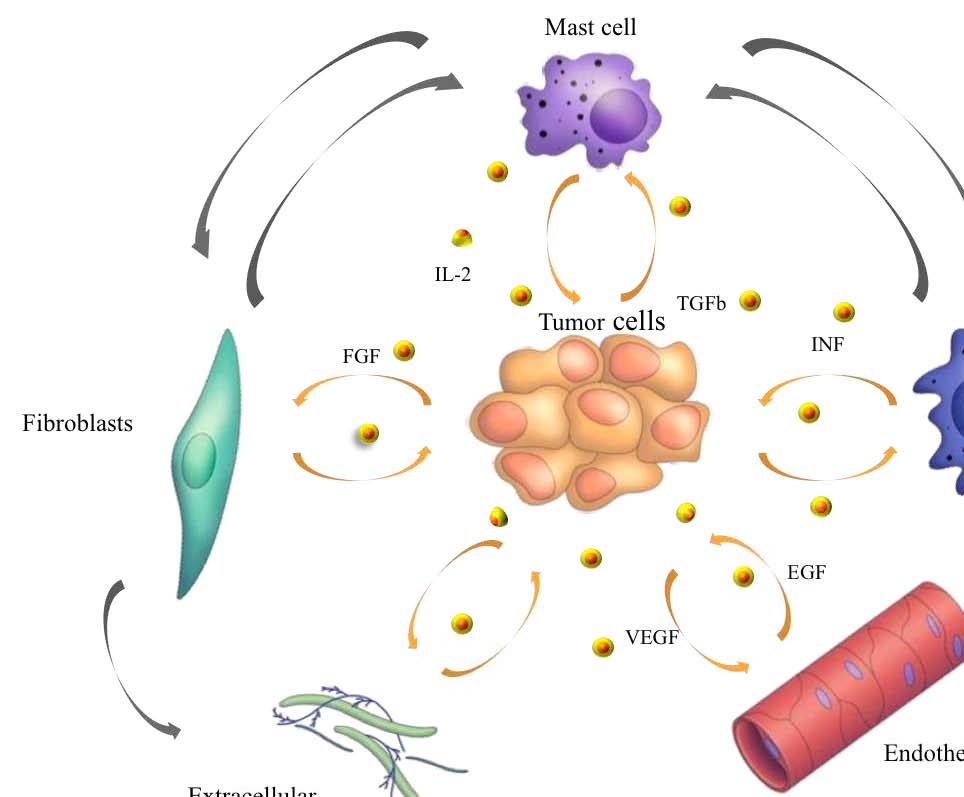
Figure 1. Tumor microenvironment. Despite their intrinsic survival and proliferation advantages,
cancer cells cannot develop into invasive cancers without reciprocal interactions with cells and soluble
mediators present in the microenvironment (Johns Hopkins Bloomberg School of Public Health).
The term myofibroblasts encompasses heterogeneous and multifunctional cell
populations exhibiting different phenotypes. Myofibroblasts were originally
described in skin wounds where they contract the stroma, bringing the epithelial
borders closer together and thereby facilitate wound healing.
Myofibroblasts modulate the stroma in physiology and pathology through direct
cell–cell contacts and through secretion of bioactive factors such as matrix
metalloproteinase (MMPs), tissue inhibitors of metalloproteinase (TIMPs),
extracellular matrix (ECM) components, growth factors, cytokines, chemokines and
lipid products (4. De Wever et al., 2008).
The formation of tumor-associated vasculature tissue is a stromal reaction essential
for tumor progression (5. Carmeliet and Jain 2000; 6. Kerbel, 2008). Tumor-
associated vessels promote tumor growth by providing oxygen and nutrients and
favor tumor metastasis by facilitating tumor cell entry into the circulation. Tumor
angiogenesis is initiated at discrete time points during tumor progression. This
angiogenic switch is determined by the balance between the genetic status of the
tumor itself, signals from stromal and recruited inflammatory cells and by the
appearance of hypoxia (7. Bergers and Benjamin 2003; 8. Coussens and Werb 2002).
Angiogenesis is a multi step process involving different cell types, in particular
endothelial, perivascular cells (i.e. pericytes, smooth muscle cells), as well as
inflammatory and stromal cells. This event is induced and regulated by secreted
growth and chemotactic factors, cell adhesion receptors, and instructive molecules.
Vascular endothelial growth factors (VEGFs) have emerged as critical mediators of
angiogenesis. The principal member of the VEGF family of factors, VEGF-A binds
to and activates two tyrosine kinase receptors, VEGFR-1 and VEGFR-2. VEGFR-2
is the major mediator of the mitogenic, angiogenic and permeability-enhancing
effects of VEGF (9. Kerbel et al. 2008).
Thus, knowledge and control of the tumor microenvironment is becoming as
important as the knowledge and control of the transformed cancer cells to better
understand cancer biology and to devise novel therapeutic approaches.
Nonetheless, information on long-distance communication between a tumor and the
host organs is still limited.
Some studies suggest that tumor cells avail special delivery systems and that a
concerted activity between tumor-derived factors and exosomes is required (10.
Albini and Sporn, 2007).
Exosomes are small multivesicular body (MVB)-derived vesicles, which are released
by tumor cells. Exosomes harbor, besides a common set of membrane and cytosolic
molecules also cell type-specific proteins that maintain functional activity. Exosomes
also contain messenger RNA (mRNA) and microRNA (miRNA) that are transferred
to target cells, where they can be translated or mediate RNA silencing. Thus
exosomal vesicles, especially cancer stem cells (CSCs)-derived exosomes, might
function as a potent tool for intercellular communication and gene delivery also in
metastasis (11. G. van Niel et al., 2006; S. 12. Keller et al., 2006).
2. Cancer Stem Cells (CSCs) and Lung
A tumor represents a population of cells with a broad spectrum of functional and
morphological heterogeneity. This phenotype could be explained by the presence of
undifferentiated "cancer stem cells" (CSCs) which represent a small subpopulation
of tumor cells in the primary tumor mass, which is responsible for tumor initiation,
growth, maintenance and spreading.
The concept of CSCs was proposed based on our current understanding of oncology
and stem cell biology. It is believed that cancer stem cells share a number of
characteristics with normal stem cells (13. Pardal et al., 2003; 14. Lobo et al., 2007).
Cancer stem cells are able to self-renew, have extensive proliferative potential and
can give rise to new phenotypically diverse progeny with variable proliferative
potential. Although the cancer stem cell hypothesis is still in a nascent stage,
increasing evidence strongly supports this model of cancer development (15. Al-Hajj
et al., 2003; 16. Singh et al., 2003; 17. Collins et al., 2005). Multiple-drug
resistance (MDR) is a property of normal stem cells that contributes to their
longevity by permitting them to survive toxic insults, including many of the drugs
currently used to treat cancer.
MDR is often mediated by overexpression of adenosine triphosphate–binding
cassette (ABC) transporters that efflux drugs out of the cell. In the absence of
specific surface antigens, this property has been used as a surrogate marker to
identify stem/progenitor cells in normal tissues, including the lung (18. Malcolm et
The conservation of these features in CSCs is the likely to be the one of the reasons
for their marked chemoresistance, which might cause the relapse often observed in
cancer patients after chemotherapeutic treatment. Apart from the similar features
between cancer stem cells and normal adult stem cells, it is essential to investigate
the differences between them in order to design new therapeutic approaches
selectively targeting CSCs while sparing the normal untransformed stem cells.
Recent studies implicate that in addition to the expression of ABC transporters CSCs
gain additional chemoresistance when they are embedded in an optimal
microenvironment (19. Malcolm et al., 2009).
This implicates that it is important to understand the crosstalk between CSCs and the
tumor microenvironment in order to fully understand the resistance mechanisms of
CSCs (Figure 2A).
Lung cancer represents the leading cause of cancer-related mortality worldwide.
Despite improvements in medical and surgical management, patient survival rates
remain stable at approximately 15%. Characterization of lung cancer stem cells
(LCSCs) is an area of active research and critical for developing novel therapies.
The presence of a clonogenic population of cells in human lung cancer was first
described more than 25 years ago. Clinical specimens from both adenocarcinoma and
small-cell lung cancer (SCLC) patients contained small populations of cells (<1,5%)
that were able to form colonies in a soft agar cloning assay. When individually
selected soft agar colonies were injected intracranially into athymic nude mice, they
were able to generate tumors with features characteristic of the original patient
specimen, thus supporting the existence of a CSC population in these malignancies
(20. Peacok and Watkins, 2008). Recent data indicates that stem cells situated
throughout the airways may initiate cancer formation. These putative stem cells
maintain pro-tumorigenic characteristics including high proliferative capacity,
multipotent differentiation, drug resistance and long lifespan when compared to
differentiated cells.
The loss of differentiation pathways could participate in maintenance of cancer stem
cells. Some studies revealed that LCSCs release soluble factors into the extracellular
compartment. These factors, called exosomes, could be able to transfer biological
information from a donor cell to a recipient one. Therefore the discovery of LCSCs
and the possibility to characterize their biological properties may provide powerful
translational tools to improve the clinical outcome of patients with lung cancer (21.
Peacock and Watkins, 2008) (Figure 2B).
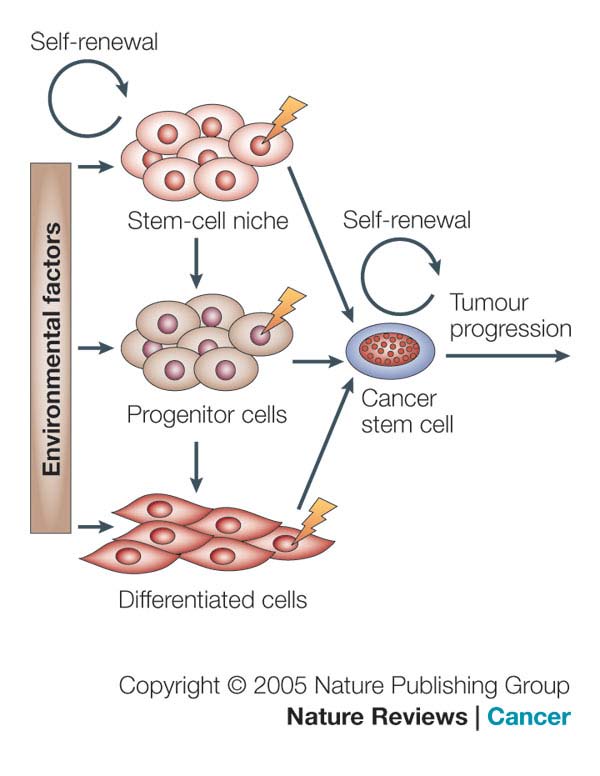
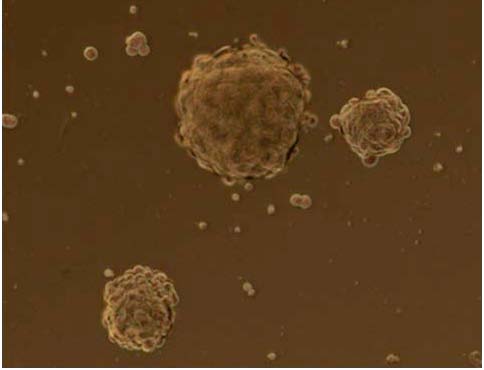
Figure 2. Cancer Stem Cells (CSCs). A) The cancer stem cell might appear after mutations in
specific stem cells or early stem cell progenitors. It is also possible that cancer stem cells can be
derived from differentiated cells. There might be numerous factors in the host microenvironment that
trigger the initial steps of tumor formation (Nature publishing group, 2005); B) Lung Cancer stem
Cells 18 (LCSCs18), a clone of our laboratory (L. Brunetto, Dept.of Hematology, Oncology and
Molecular Medicine at Istituto Superiore di Sanità, Rome).
3. CD133 stem cell marker
In recent years it was described that solid tumor contain cancer stem cells. These
tumor-initiating cells often display characteristic of chemotherapy resistance. CSCs
can drive tumor growth and possess multilineage differentiation potential, allowing
them to reform the original malignancy on transplantation in mice.
In several cancers, CSCs have been identified using one or multiple markers, like
CD24, CD29, CD44, CD133, ALDH1, or Hoechst exclusion. The pentaspan
membrane protein CD133, originally identified as a marker for CD34+ hematopoietic
stem and progenitor cells, has been used for CSC identification in several types of
cancer, such as glioblastoma, melanoma, liver cancer, lung cancer, osteosarcoma,
and colon cancer (22. Kemper et al., 2010). Mizrak et al. highlighted also the role of
CD133 as a marker of CSCs in various human tumors (Table 1). CD133 was
expressed in combination with CD44+ and α hi
in approximately 0,1% of cells
within a large series of prostate tumors, irrespective of their grade or metastatic state.
These cells were capable of self-renewal, proliferation, and multi-lineage
differentiation in vitro to recapitulate the original tumor phenotype, consistent with
CSC properties (23. Mizrak et al., 2008).
Antigenic phenotype
CD133, CD44, α1β2
Human prostate tumors
Human pancreatic adenocarcinoma
Human colon carcinoma
Human hepatocellular carcinoma
Human neural carcinoma
Human renal carcinoma
Table 1. Cells expressing CD133, often in combination with other markers, represent cancer stem
cells in a variety of human tumors (Mizrak et al., 2008).
Olempska et al. (24) showed that ABCG2 and CD133 co-expression was elevated in
two out of five human pancreatic adenocarcinoma cell lines, and thus CD133 may
also represent a putative CSC marker in the pancreas. CSCs are significantly
enriched in CD133 subpopulations derived from human colon cancer and
hepatocellular carcinomas, as shown by their potential to both self-renew and
differentiate, to form colonies and proliferate in vitro, and by their ability to reform
the original tumor phenotype when transplanted either subcutaneously or into the
renal capsule of immunodeficient mice. CSCs expressing CD133 and nestin were
isolated from several human brain tumours, including medulloblastomas,
glioblastomas, and oligoastrocytomas. The human CD133 protein is encoded by the
PROM1 gene on chromosome 4p15 and codes for a five-transmembrane
Human CD133, also known as AC133 and prominin-1, contains 5 transmembrane
domains and 2 large glycosylated extracellular loops (25. Wu and Wu, 2009); the
predicted size of CD133 is 97 kDa, but the actual molecular weight of glycosylated
CD133 is 120 kDa (Figure 3).
Figure 3. CD133 protein structure. CD133 protein has an extracellular N-terminus, 5 hydrophobic
transmembrane domains, 2 small cytoplasmic loops, 2 large extracellular loops and a cytoplasmic C-
termus (L. Brunetto).
CD133 localizes to plasma membrane protrusions at the apical surface of cells,
reflecting a polarized cell structure. Several monoclonal antibodies have been
developed against CD133. The most commonly used are AC133 (CD133/1) and
293C/AC141 (CD133/2), which are reported to recognize distinct epitopes (26.
Missol-Kolka et al., 2010). AC133 is frequently used to isolate CSCs and suggested
to recognize a glycosylated epitope on CD133, which contains eight putative N-
linked glycosylation sites. Several groups have shown that AC133+, but not AC133-,
cells sorted from primary solid cancers can form tumors in immunodeficient mice
that recapitulate the morphology of the original tumor (27. Kemper et al., 2010).
The specific localization of CD133 may suggest that CD133 is involved in
organization of plasma membrane protrusions. Interactions between CD133 and
cholesterol within such novel membrane micro-domains suggested that CD133 might
also be important in maintaining an appropriate lipid composition within the plasma
membrane (28. Mizrak et al., 2008). CD133 shows homologies with proteins
involved in plasma membrane fusion especially in endocytic pathway, but its
function is still unclear. Similar to CD133, prominin-2 is selectively expressed in
association with cholesterol, within microdomains of epithelial plasmalemmal
protrusions, and is released from small vesicles and found in physiological fluids (29.
Corbeil et al., 2010).
However, in contrast to the polarized apical membrane location of CD133, prominin-
2 shows a non-polar distribution between the apical and basolateral membranes of
epithelial cells (30. Mizrak et al., 2008). Due to its expression in different kinds of
CSCs, CD133 is a marker of big interest in order to better understand the biological
properties and the mechanism of resistance of CSCs, in order to develop targeted
4. CSC-derived Exosomes
Exosomes are small natural membrane vesicles released from cells and have been
subject of intensive research in recent years. Multivesicular bodies (MVBs) and their
intraluminal vesicles (ILVs) are involved in the sequestration of proteins destined for
degradation in lysosomes. An alternative fate of MVBs is their exocytic fusion with
the plasma membrane leading to the release of the 50–90 nm ILVs into the
extracellular milieu. The secreted ILVs are then called exosomes (31. G. van Niel et
al., 2006). After their initial description as vesicles of endosomal origin and secreted
by reticulocytes during differentiation, vesicles with the hallmarks of exosomes
appeared to be released by other cells. Exosomes are present in the culture
supernatant of several cell types of hematopoietic origin (B cells, dendritic cells,
mast cells, T cells and platelets) and of non-hematopoietic origin (intestinal epithelial
cells, tumor cells, Schwann cells and neuronal cells) (32. G. van Niel et al., 2006).
In addition there is increasing evidence for the presence of exosomes in
physiological fluids such as plasma, malignant and pleural effusions and urine (33.
G. van Niel et al., 2006; 34. S. Keller et al., 2006). The lipid and protein content of
exosomes has been extensively analyzed by various techniques including Western
blotting, fluorescence-activated cell sorting (FACS), immune electron microscopy
(immuno-EM) and mass spectrometry. The protein and lipid composition of
exosomes varies depending on the cell type of origin. Nevertheless, exosomes
contain a number of common protein components. The cytosolic proteins present on
exosomes include Rabs, which promote exosome docking, and the membrane fusion
events and annexins (annexin I, II, V and VI) that may regulate membrane
cytoskeleton dynamics. Also several adhesion molecules such as intercellular
adhesion molecule-1, CD146, CD9, CD18, CD11a, CD11b, CD11c, CD166 and
LFA- 3/CD58 have been identified in exosomal preparations (35. Taylor and Taylor
Exosomes also contain heatshock proteins such as Hsp70 and Hsp90 and a
characteristic presence of tetraspanins, which include CD9, CD63, CD81 and CD82.
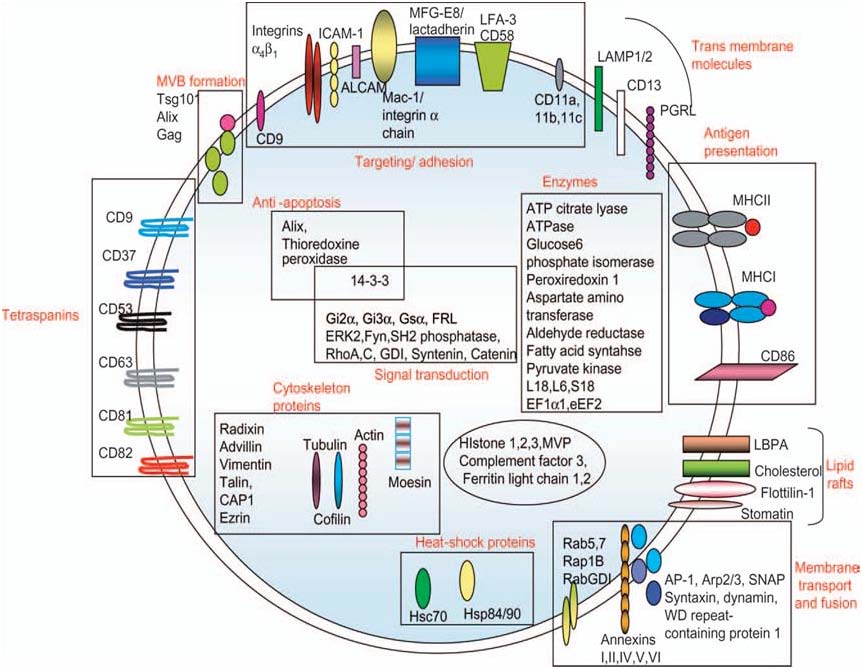
But exosomes also carry some cell-specific proteins like MHCII and CD86 present
only on exosomes isolated from antigen-presenting cells (APCs) (36. Morelli et al.,
Due to their endosomal origin exosomes are enriched in proteins that participate in
vesicle formation and trafficking like the lysobisphosphatidic acid (LBPA)-binding
protein Alix. Other proteins detected on exosomes are the metabolic enzymes such as
peroxidases, pyruvate and lipid kinases and enolase-1 (37. Jeffrey S. Schorey and
Sanchita Bhatnagar, 2008) (Figure 4).
Figure 4. Protein composition of exosomes. The names, location and functions are indicated (i.e.
membrane bound or soluble) and in some cases their function. GDI, GTP dissociation inhibitor;
ICAM1, intercellular adhesion molecule-1; CAP-1, adenylyl cyclase-associated protein; LAMP,
lysosomal associated membrane protein-1; PGRL, PG regulatory-like protein (Schorey and
Bhatnagar, 2008).
The exact molecular composition of exosomes reflects the specialized functions of
their original cells. Through their ability to bind target cells, they are likely to
modulate selected cellular activities, such as vascular homeostasis and antigen
presentation. The presence of exosomes in blood and tissues in vivo suggests their
participation in physiological and/or pathological processes. Several studies have
analyzed the biological activities of exosomes in vitro, but very little is known about
their possible functions in vivo (38. Thery, 2011). Therefore, secreted exosomes are
biologically active entities, which might be important for the induction of variety of
pathways, including those are responsible for a tumor progression.
Tumor cells, as well cancer stem cells (CSCs), release these vesicular structures in
the tumor microenvironment, carrying a large array of proteins from their originating
cells. CSCs express on their surface CD133 protein; this marker seems also to be
present on CSCs-derived exosomes (39. Costas et al., 2009). Even if CD133 is
considered a stem cell marker, it is important to investigate a potential role of this
protein in the tumor microenvironment in order to understand the crosstalk between
CSCs and the tumor microenvironment and the resistance mechanisms of CSCs.
Knowledge and control of the tumor microenvironment is becoming as important as
the knowledge and control of the transformed cancer cells to better understand
cancer biology and to devise novel therapeutic approaches.
5. microRNAs
The hallmark of a stem cell is its ability to self-renew and to produce numerous
differentiated cells. This unique property is controlled by dynamic interplays
between extrinsic signaling, epigenetic, transcriptional and post-transcriptional
regulations. Recent research indicates that microRNAs (miRNAs) have an important
role in regulating stem cell self-renewal and differentiation by repressing the
translation of selected messengerRNAs (mRNAs) in stem cells and differentiating
daughter cells. miRNAs are 20–25 nucleotide (nt) non-coding RNAs that bind to the
3' untranslated region (3'UTR) of target mRNAs through an imperfect match to
repress their translation and stability. This is achieved by forming a
ribonucleoprotein complex, called the RNA-induced silencing complex (RISC).
Occasionally, miRNAs have also been observed to activate target mRNA translation
and to regulate their stability.
miRNAs are derived from precursor transcripts, called primary miRNAs (pri-
miRNAs), that are first processed in the nucleus into an intermediate form (pre-
miRNAs) by the Microprocessor protein complex. The pre-miRNAs are then
translocated by the exportin 5–RanGTP shuttle system into the cytoplasm, in which
they are further processed by Dicer, an RNase III-like enzyme, into mature miRNAs
(Figure 5), (40. Gangaraju and Lin, 2009).
Expression of approximately 30% of human proteins appears to be regulated by
miRNAs. Through interactions with 3'UTRs, miRNAs can modulate the expression
of many genes simultaneously, often regulating individual signaling pathways at
multiple levels (41. Hatley et al., 2010).
An integral role of miRNAs in cancer pathogenesis has begun to emerge. miRNA
expression profiling reveals characteristic signatures for many tumor types, including
non-small-cell lung cancer (NSCLC) and are predictive of tumor classification,
prognosis, and response to therapy (42. Calin and Croce, 2006).
miRNA expression patterns are remarkably reliable markers of cancers; in some
cases, they have even proven more reliable than conventional histology.
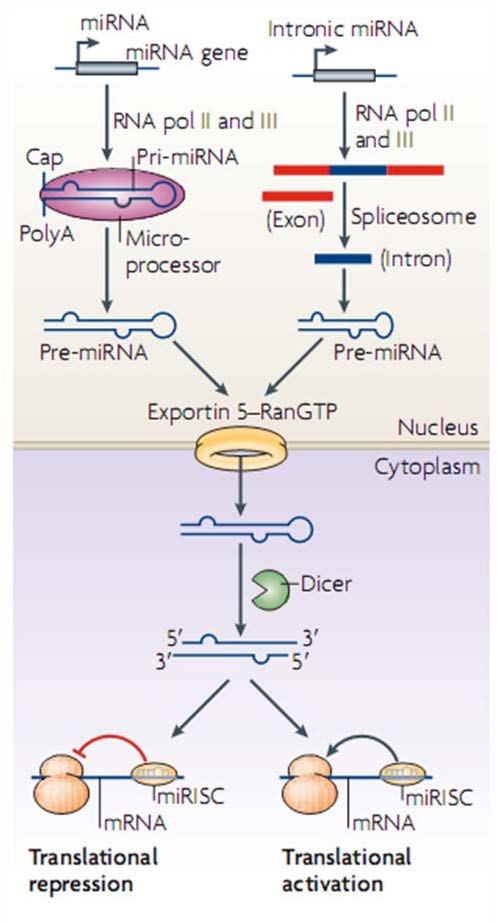
Figure 5. Biogenesis and regulatory features of the miRNA. MicroRNA (miRNA) genes are
transcribed by RNA polymerases II and III into primary transcripts called the pri-miRNAs. These are
processed into pre-miRNAs in the nucleus by a Microprocessor complex. Pre-miRNAs are then
transported by exportin 5, which is a karyopherin, and RanGTP into the cytoplasm, where they are
further processed by the RNAse III enzyme Dicer. This results in double-stranded 20–25 nucleotide
(nt) intermediates with 2 nt overhangs on the 3' end (Gangaraju and Lin, 2009).
miRNAs are capable of functioning as classical tumor suppressors or oncogenes,
thus actively participating in human cancer pathogenesis (43. Hatley et al., 2010).
For example a miRNA recently well studied is miR21; a large-scale survey to
determine the miRNA signature of 540 tumor samples, including lung, breast,
stomach, prostate, colon, and pancreatic tumors and their respective normal adjacent
tissue, revealed miR-21 was the only miRNA upregulated in all these tumors.
Functional studies suggest that miR-21 has oncogenic activity. Indeed, knockdown
of miR-21 in cancer cell lines activates caspases leading to apoptotic cell death,
suggesting miR-21 is an antiapoptotic factor (44. Hatley et al., 2010). Growing
evidence suggests that extracellular microRNAs (miRNAs) stably exist in human
body fluids, including plasma, saliva and urine, although ribonucleases (RNases) also
circulate throughout the body. This finding indicates that miRNAs are excreted after
they are contained in RNase-resistant lipid vesicles, such as exosomes and apoptotic
bodies. However, the secretory mechanism and biological function of extracellular
miRNAs remain unclear. Recent study demonstrated that the secretion of miRNAs is
triggered by the elevation of the cellular amount of ceramide, which is a bioactive
sphingolipid, whose synthesis is tightly regulated by neutral sphingomyelinase 2
(nSMase2) (Figure 6, 45. Iguchi et al., 2010).
Figure 6. Ceramide-dependent secretory pathway. A working model of secretory mechanism of
microRNAs. Multivesicular bodies (mVBs) are an important cellular compartment for the metabolism
of proteins and miRNAs. Ubiquitinated proteins are incorporated into lysosomes via endosomal
sorting complex required for transport (eSCrt) machinery, subsequently followed by degradation or
excretion. miRNAs are packaged into exosomes and the release from the cells is stimulated by the
surge of cellular ceramide (Iguchi et al., 2010).
Gibbings et al. (46) also showed that RNA extracted from secreted vesicles that
resemble exosomes (50-100 nm in diameter) contains miRNAs. These exosomes
derive from endo-lysosomal compartments called multivesicular bodies (MVBs),
which seem modulate miRNA activity.
Currently, accumulating evidence suggests that exosomal vesicles can function as
intercellular transmitters to convey their contents, in particular, microRNA
(miRNA). Recent studies reported that extracellular exosomal miRNAs were
transferred into other cells and that apoptotic bodies delivered miR-126 into
endothelial cells (47. Iguchi et al., 2010). Despite these advances, however, the
underlying mechanism of the secretory process and the biological function of
circulating miRNAs are not yet fully understood. Interestingly, the amounts of
secretory miRNAs are upregulated in the plasma of patients bearing tumors,
including B cell lymphoma, prostate cancer, lung cancer, and ovarian cancer.
miRNAs secreted from donor cells can be taken up and function in the recipient
These findings propose a novel mechanism of intercellular communication mediated
by secretory miRNAs. Thus, detection and monitoring of tumors are now becoming
possible by the evaluation of tumor-derived secretory miRNAs (48. Kosaka et al.,
Working Hypothesis
The aim of this project is to investigate the biological cross talk between CSCs and
the tumor microenvironment. This interchange of informations may occur through
CSC-derived exosomes released into tumor microenvironment (Figure 7).
Figure 7. Working Hypothesis. Cancer Stem cells (CSCs) release in the tumor microenvironment the
exosomes, which have similar characteristics of origin cells. Exosomes are bioactive molecules, which
could stimulate target cells to develop and/or expand tumor capacities such as growth,
migratory/invasive potential and chemoresistance (L. Brunetto).
Exosomes contain a characteristic composition of proteins, a variety of
messengerRNAs and microRNAs. Moreover exosomes express on their surface cell
recognition molecules that facilitate their selective targeting and uptake by recipient
cells. We suggest that exosomal secretion of proteins, such as CD133 and
microRNAs may be a fundamental mode of communication between cells of tumor
microenvironment. In order to investigate the exosome-mediated crosstalk between
CSCs and their microenvironment we will set up a isolation procedure to obtain
exosomal particles from CSCs of solid tumors such as lung carcinoma. Having
established the experimental system to isolate exosomes, we will characterize the
biochemical properties of these vesicles. Moreover the influence of exosomes on
differentiated tumor cell lines and also on non-tumoral cells such as fibroblast will be
investigated. We will study also exosomes properties released in the supernatant of
stable Prominin-1 transduced cell lines. Furthermore, we will incubate non-
transformed target cells in exosome-containing medium and investigate changes in
their mRNA expression to define intracellular pathways activated by CSC derived
exosomes. In addition we will analyze the lysates of exosome-treated cells for
changes in their phosphoproteomic profile. This will provide us with additional
information, which intracellular pathways are activated that not necessarily lead to a
marked change in the gene expression profile. We will investigate how CD133
protein is localized on exosomal vesicles and if this key protein has a functional role,
such as fusion of membranes, on target cells. Therefore we will perform functional
assay, orientation and uptake kinetic experiments in order to partially extend our
knowledge about CD133 stem cell marker. The treatment of tumor cells with
exosomal particles, which are released into the supernatant, will be a very useful
experimental strategy to understand if the exosomes have the capacity to influence
target cells. Exosomes released in the supernatant are bioactive molecules and they
could activate significant cellular pathways. Therefore exosomal vesicles could
stimulate target cells in order to develop tumor capacities such as migration and
Materials and Methods
1. Lung Cancer Stem lines
In this study we worked with different lung cancer stem lines, especially with the
clones LCSC18, LCSC34 and LCSC136, obtained from Dott. Adriana Eramo. These
clones were isolated from lung carcinoma tissue specimens. Tissues were
mechanically dissociated with sterile scissors and forceps in DMEM (Dulbecco's
Modified Eagle Medium, Gibco Invitrogen Inc., BRL, Rockville, MD) containing
Streptomycin 500g/mL, Penicillin 500U/mL (PAA Laboratories Inc.) and
Amphotericin-B 5g/mL (Fungizone, Gibco). Then tissues were dissociated
enzymatically at 37°C using type II collagenase 1.5mg/mL (Gibco Invitrogen Inc.)
DNase I 20g/mL (Roche, Mannheim, Germany) for a time depending on piece size.
Cell suspension was then passed through a sterile 100mm filter. Following a further
series of washes in PBS cells were then placed in culture in a DMEM-F12
(Hepes15mM L-glutamine, Invitrogen Life Technologies Inc., Grand Island, NY)
containing medium and supplemented with b-FGF (Fibroblastic Growth Factor type
II) 10ng/mL and EGF (Epidermal Growth Factor) 20ng/mL (PeproTech, Rocky Hill,
NJ). Culture medium also contains 10% of Hormone mix (composed by 500mg
insulin, 96,6mg putrescine, 1g apotransferrin, 200mlDMEMF12 5X, 20ml glucose
30%, 5ml Hepes, from Sigma). Culture medium was replaced twice a week until the
formation of spheroids in suspension was observed. Spheroids were passaged by
mechanical dissociation followed by replating of individual cells and small
aggregates in fresh medium. LCSCs were incubated at 37°C with 5% CO2. For the
cultivation of the LCSC clones we used flasks, which were not treated for tissue
culture to reduce cell adherence and support the growth as undifferentiated tumor
spheres. The serum free stem cell medium was supplemented with fresh growth
factors to ensure the maintenance of the cells as floating aggregates. Cultures were
expanded by mechanical dissociation of spheres, followed by re-plating of both
single cells and residual small aggregates in complete fresh medium. Growing them
in a different culture medium, which induces their differentiation, we tested the
accuracy of LCSCs stemness.
2. Adherent Cell lines
In our experiments we used, also, several adherent cell lines; the HEK293T human
renal epithelial cell line (CRL-11268 ATCC Number) were maintained in
Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal
bovine serum (FBS), they were incubated at 37°C and 5% CO2. They are extremely
easy to work with, being straightforward to culture and to transfect, and so can be
used in experiments in which the behavior of the cell itself is not of interest.
Typically, these experiments involve transfecting in a gene of interest, and then
analyzing the expressed protein. The widespread use of this cell line is due to its
extreme transfectability by the various techniques, including calcium phosphate
method, achieving efficiencies approaching 100%. The NIH3T3 mouse embryonic
fibroblast cell line (CRL-1658 ATCC Number) was used in our phospho-proteomic
analysis; these cells were also maintained in DMEM, 10% FBS, at 37°C and 5%
CO2. The original cells are extremely contact inhibited, although the cell line is no longer inhibited; NIH3T3 cells are sensitive to sarcoma virus focus formation, as
well as leukemia virus. NIH3T3 mouse cells are hypertriploid and the modal
chromosome number is 68, which occurs in 30% of cells.
The HCT116 colorectal carcinoma cell line (CCL-247 ATCC Number) were used in
our experiments in order to setup the tetracycline-dependent regulatory system;
therefore the cells were stable transduced with two different vectors: TetOGFP and
rtTA3GFP ones. The cells were grown in McCoy's 5a Medium (GIBCO), 10% FBS,
100 units/ml penicillin, 100 µg/ml streptomycin (GIBCO). If 5x106 cells are plated
onto a 75cm2 flask, the culture typically reaches 70-90% confluence in 2-3 days and
is ready to split. The A549 adenocarcinomic human alveolar basal epithelial cells
(CCL-185 ATCC Number) were maintained in DMEM, supplemented with 10%
FBS and incubated at 37°C and 5% CO2. These cells are squamous in nature and responsible for the diffusion of substances, such as water and electrolytes, across the
alveoli of lungs. They grow adherently, as a monolayer, in vivo. The A549 cells are
positive for keratin, as is evidenced by immunoperoxidase staining.
These cells are also able to synthesize lecithin and contain a high percentage of
desaturated fatty acids, which are utilized by the cytidine-diphospho-choline pathway
and important for the maintenance of membrane phospholipids in cells. Also these
cells were used in the tetracycline-dependent regulatory system experiments and
transduced with the vectors TetOGFP and rtTA3GFP.
3. Virus production and Infection
Transfection of DNA in mammalian cells is commonly used to study the regulation
and functions of genes, as well as to express proteins and produce viruses. To
produce the virus, all three constructs are simultaneously transfected into HEK293T
host cells and the culture supernatant containing infectious viral particles is
subsequently harvested. For virus particles production we used HEK293T cells and
performed the calcium phosphate method whit maximum efficiency and minimal
toxicity. We thaw fresh HEK293T cells and put them in culture. The day before
transfection HEK293T cells were seeded in a T25 flask. The host cells were
cotransfected with 6µg pMD2 vector, 14µg psPAX2 and 20µg DNA of interest (such
as pTWProm1GFP, pTWGFP, TetO-CMV-Luc-GFP, rtTA3-GFP vectors) in 120µl
CaCl2. The transfection mixture was filled until 1ml with pure water and we added drop by drop 1ml 2XHBS, bubbling. Virus-containing supernatant was collected 72h
post transfection. It was centrifuged for 5 minutes at 1400rpm, filtered with 0,45µm
filter and incubated with cells (e.g. HCT116, HEK293T, LCSC136) to infect with, in
presence of polybrene (hexadimethrine bromide, 1:1000), which is a cationic
polymer used to increase the efficiency of infection. The cells incubated with virus
were centrifuged for 45 minutes at 1800rpm and put in the incubator for 1h; the
infection procedure was repeated two times. Transfection efficiency was analyzed
same days post transduction; the cells were analyzed for GFP expression using
fluorescence microscope and by FACS analysis.
4. Isolation of Lung Cancer Stem Cells (LCSCs)-derived exosomes
Exosomes possess a defined set of membrane and cytosolic proteins. The
physiological function of exosomes is still a matter of debate, but increasing results
in various experimental systems suggest their involvement in multiple biological
processes. Because both cell culture supernatants and biological fluids contain
different types of lipid membranes, it is critical to perform a high-quality exosome
purification. Large scale exosome isolation procedure is well established in literature.
In order to obtain exosomes from different LCSCs, the isolation protocol was based
on the published protocol, but to reach high yields, we had to include slight
modifications. Filtration/ultracentrifugation steps were used to purify exosomal
vesicles. Briefly, human LCSCs were grown to confluence in serum free LCSC
medium. Supernatants from LCSC cultures were harvested, centrifuged for 10
minutes at 300g, for 30 minutes at 3000g (Beckman-Coulter centrifuge) and filtered
through a 0.22µm sterile filter (Millipore) to eliminate impurities such as large
cellular debris. Next step was to concentrate supernatants using Vivaspin 20
centrifugal concentrator MWCO300000 (Sigma-Aldrich). The supernatants were
then ultracentrifuged for 1h at 150,000g (Beckman-Coulter L-90K ultracentrifuge,
SW41 rotor). The pellets were washed by resuspension in 11ml PBS and collected by
ultracentrifugation for 1h at 150,000g (Figure 8). Finally the pellets, containing the
exosomes, were resuspended in a small volume (50-100µl) of PBS. The exosomal
protein was quantified using the Biorad protein determination kit with BSA as a
standard. We measured 10µl exosomes preparation adding 1 µl NP40 2%, on ice 5'.
Then we took 10 µl in 150 µl Bradford for the measurement. The exosomes were
kept in aliquots at -80°C for functional assay and western blot analysis.
Figure 8. Exosomes isolation protocol. The isolation procedure to obtain exosomal particles from
the supernatant of LCSCs cultures is characterized by differential centrifugation steps, as described
above (L.Brunetto).
5. Western Blot analysis
The western blot technique was used to detect specific proteins from purified
exosomes, immunoprecipitation samples and cell lysates, in order to detect
respectively: specific exosomal markers, the correlation between the presence of
specific exosomal markers and CD133 protein and the activation of phospho-
proteomic cellular pathways. An equal concentration of proteins from each samples
was subjected to electrophoresis on acrylamide gel containing SDS (Sodium Dodecyl
Sulfate). NuPage Novex Bis-Tris gel 1.0 mm x 12 well 4-12% (Gibco Invitrogen
Inc.) was used. An equivalent of 20 µg of protein, supplemented with loading buffer
NuPage LDS sample buffer (25mM Tris-HCl, pH 6.8, SDS 10%, 50% glycerol, 5%
β-mercaptoethanol, 0.01% bromophenol blue, Gibco Invitrogen Inc.) was incubated
at 75°C for 10 minutes and loaded. SeeBlue Plus2 (Gibco Invitrogen Inc.) was used
as molecular weight marker. Electrophoresis was performed in MES or MOPS
running buffer at 125 V for 10 minutes and 180V for 50 minutes. Then we
transferred the gel onto nitrocellulose filters (Amersham). Proteins transfer was made
by using an elettroblot (Invitrogen) filled with transfer buffer (NuPage Transfer
Buffer 20X Invitrogen and 20% methanol) at 30 V for 2 hours. Membrane was
saturated using PBST (Phosphate buffered saline, 0.2% Tween-20) containing 5%
powder milk (Blottin-Grade Blocker non- fat dry milk, Bio-Rad). After saturating the
excess of protein bindings sites on the membrane, the filters were incubated
overnight at +4°C with the primary antibodies. The primary antibodies were diluted
in PBST. The following day the filters were washed three times with PBST on the
shaker. All the filters were normalized by actin (1:10000 Sigma) or by another
ubiquitin protein on the basis of the analysis. Specific antibodies were used in this
analysis in order to detect specific markers of LCSC-derived exosomes: tumor
susceptibility gene TSG101 (1:1000 Abcam), transferrin receptor CD71 (1:500,
Santa Cruz), tetraspanin CD9 (1:500, Santa Cruz), member Ras oncogene family
Rab5b (1:500 Santa Cruz), and stem cell marker CD133 (1:800 Abcam).
In order to detect the phosphorylation status of recipient cells, after the incubation
with CD133 enriched SN, we used following antibodies: phospho-ERK (1:1000,
Santa Cruz), phospho-p38 (1:1000, Cell Signaling), phospho-AKT (1:1000, Cell
Signaling), phospho-GSK3β (1:1000, Cell Signaling), ERK1 (1:1000, Santa Cruz),
AKT1 (1:500, Santa Cruz).
The secondary antibodies used were: anti-Rabbit HRP-linked 1:40.000, anti-Mouse
IgG1 HRP-linked 1:20.000, anti-Mouse IgG2a HRP-linked 1:20.000, anti-Goat
HRP-linked 1:30.000, from Amersham. The secondary antibodies were diluted in
PBST with a little addition of PBSTmilk. After the appropriate secondary-HRP
conjugated antibodies incubations, protein signals were visualized by
chemiluminescence (Pierce Super Signal West Pico).
6. FACS analysis
The FACS analysis was performed using BD FACS Canto machine. Flow cytometry
was performed to verify different aspects in our experiments. For example we used
this analysis to distinguish the LCSCs stemness status from the differentiated one;
subsequently a virus infection we tested the stable transduction of a cell line by GFP
FACS analysis; we verified the markers profile of the cell lines used; after uptake
experiments and kinetic ones we were able to verify the presence of CD133 protein
on recipient cells and the timing of uptake respectively using CD133-PE antibody
from Miltenyi (1:20); for our aims, we sorted by FACS cells of interests from the
negative ones. For FACS analysis we used the antibodies: CD81 (1:50 Santa Cruz),
CD9 (1:50 Santa Cruz).
7. Immunoprecipitation (IP)
Immunoprecipitation (IP) is a technique of precipitating a protein antigen out of
heterogeneous solution using an antibody that specifically binds to that particular
protein. This process can be used to isolate and concentrate a protein from a sample
containing many thousands of different proteins. The antibody-protein complexes are
precipitated from the solution by adding of specific beads binding the complexes
(Figure 9). We used IP technique in order to extract from CD133 enriched SN a
specific component identifiable as exosomal vesicles, using the biotinylated CD133
antibody (0,5µg for each sample) from Miltentyi. In our experiments we used
Streptavidin-coupled Dynabeads from Invitrogen, which are the gold standard for
isolation and handling of biotinylated nucleic acids, antibodies or other biotinylated
ligands and targets. The very high binding affinity of the streptavidin-biotin
interaction is utilized in a vast number of applications. These uniform and
superparamagnetic beads are 1 µm in diameter, with a monolayer of recombinant
streptavidin covalently coupled to the surface and further blocked with BSA.
Dynabeads are supplied in PBS, pH 7.4 with 0.1% BSA and 0.02% NaN3 added as
preservatives. We performed IP from supernatants of HEK293TGFP (as a control)
and HEK293TProm1GFP cell lines. We harvested 2ml of each supernatants and
centrifuged them for 5' at 1400rpm. We incubated the supernatants with biotinylated
CD133 antibody and with biotinylated IgG control antibody for 2h on the wheel at
room temperature. Then we added in each sample 50µl of Streptavidin beads,
previously washed three times with 1ml PBS, for other 2h on the wheel. Then we
pelleted the beads and washed them three times with 1ml PBS. The sample beads
were ready for the WB analysis.
Figure 9. Immunoprecipitation (IP) assay. The IP procedure is characterized by different steps: l)
adding Antibody against protein of interest (CD133); 2) Antibody binds to protein of interest; 3)
adding Streptavidin magnetic beads make antibody-protein complex insoluble; 4) centrifugation of
solution pellets the complex of interest; (L.Brunetto).
8. Screening for optimal cellular system to investigate the uptake of
CD133 containing particles
In order to detect the best conditions for uptake experiment we tested different cell
lines as recipient cells and supernatant preparations. Therefore we performed
screening experiments in order to investigate the potential uptake of exosomes by
recipient cells. We incubated A549 lung cell line with LCSC18 and with
HEK293TProm1GFP supernatants. Both yellow supernatants were harvested after
some days in culture and were incubated with A549 cells for 12h. The following day
A549 cells were harvest and stained for CD133 (1:20, Miltenyi) for 30'on ice; we
used as secondary antibody anti-mouse-PE (1:100, Sigma) for 20' on ice. A549 cells,
treated with two kinds of supernatants, were analyzed by FACS. We didn't see in
A549 cells any CD133 signal of uptake. Therefore we tried to improve our uptake
experiment changing recipient cell line and supernatant conditions. We used
HEK293T cells, which were incubated with CD133 enriched supernatant from
LCSC18. Before incubation the supernatant was treated in two ways; supernatant
was ultracentrifuged at 33.000rpm for 2h; then ultracentrifuged pellet was
resuspended in DMEM medium and put on HEK293T cells for 12h. LCSC18 was
treated also in another way: it was concentrated using Vivaspin 300.000 Da, from
Sigma-Aldrich, starting from 40ml of supernatant; then this supernatant was also put
on HEK293T cells for 12h. The following day HEK293T cells were harvest and
stained for CD133 (1:20, Miltenyi) for 30'on ice; we used as secondary antibody
anti-mouse-PE (1:100, Sigma) for 20' on ice. By FACS HEK293T cells, treated with
two supernatant preparations, were analyzed (see 4. Results section).
9. Exosomes uptake assay
Established the ideal experimental conditions, finding as appropriate recipient cell
line the HEK293T cells, we performed exosomes uptake experiments. We first grew
HEK293T cells on the slides. Then we put on these recipient cells concentrated
supernatant (3-5 fold) from HEK293TProm1GFP stable transduced cell line or from
LCSC18 for 30', 60', 120' and 240' at 37°C. We then washed the HEK293T cells
with PBS and fix them in 2%PFA at 4°C for at least 1h. Then cells were stained for
immunofluorescence and flow cytometric analysis using specific CD133 antibody to
verify neo-positive cells for CD133 after the incubation. In order to quantify the
cellular uptake of exosomes, the experiment was repeated four times. For
immunofluorescence all the settings of imaging and processing were kept constant
and the relative fluorescent intensities were calculated.
The same experimental procedure was performed in uptake kinetic experiments, in
order to calculate how long CD133 is kept into the HEK293T cells.
We first grew HEK293TGFP (control cell line) and HEK293TProm1GFP (CD133
cell line) stable transduced cell lines in order to harvest a supernatant enriched in
exosomes; then we concentrated these two supernatants. In this experiment the
control and the CD133-enriched supernatant were maintained on the recipient cells
over night at 37°C. The following day both supernatants were washed away with
PBS at different time points. HEK293T cells were fixed in 2%PFA at 4°C for at least
1h and then stained for immunofluorescence and flow cytometry analysis.
10. Immunofluorescence (IF)
As we did by FACS analysis, we used Immunofluorescence technique to
demonstrate that exosomal vesicles, contained in the supernatant, are able to transfer
CD133 protein from expressing to non-expressing cells. In order to visualize this
specific protein transfer, first the cells were grown on cover slips. After the
incubation with concentrated CD133-enriched supernatant we permeabilized the
samples with PBS/Triton 0.5%, 15' at RT and then we incubated the slides overnight
with CD133/2 (AC141) antibody (1:10, Miltenyi) at +4°C. The following day we
washed the samples three times with PBS BSA 3% and we stained them for DAPI
(10ug/mL), Phalloidin-Alexa647 (0.2U/µL) and anti-Mouse Alexa555 (2ug/mL) for
2h at RT. Finally the slides were closed using the Vectashield mounting medium. All
preparations were examined under an Olympus FluoView FV1000 Confocal
Microscope using a 60X 1.35 N.A lens. Images were acquired with the same
confocal microscope using FV1000 software.
11. Transfection procedures
The process of introducing nucleic acids into eukaryotic cells by non-viral methods
is defined as transfection. For our experiments we used as transfection reagent the
FuGENE® 6 Transfection Reagent (by Roche). FuGENE is a multi-component
reagent that forms a complex with DNA and transports it into animal cells. FuGENE
has several advantages, such as high transfection efficiency in many common cell
types, no cytotoxicity, suitable for transient and stable transfection, it functions well
in the presence or absence of serum. The recipient cells (e.g. HCT116, HEK293T)
were seeded in 12wellplate and for each well we prepared: 50µl Optimem medium
(Gibco 1X) with 3µl/1µg DNA and we incubated for 5 minutes at room temperature.
Then we added 200ng DNA of interest and incubated for 15 minutes at room
temperature. The transfection mix was added on the recipient cells.
We used another transfection method that is the Calcium Phosphate transfection
method. This works best in cell lines that are highly transformed and adherent.
In this case the transfection efficiency depending closely on the cell line used. The
method works well for transient experiments but very well for generating stable cell
lines (see 3. of Materials and Methods).
12. QReal Time RT-PCR
For RNA analysis, total RNA was purified with TRIZOL Reagent (Invitrogen),
following the manufacturer's instructions. Quantitative analysis of miRNAs of
interest was carried out on RNA samples by Real-time PCR.
Reverse transcription was performed using microRNA-specific primers (5x primer,
Applied Biosystems, Foster City, CA), and Superscript II enzyme (Invitrogen). The
reaction mix has a finale volume of 15µl and the following composition:
• Buffer SuperscriptII 1x
• RT primer 1x
• Superscript II (30 U)
• RNA (for exosomes samples 50 ng )
• 30' a 42°C (Superscript at 42°C) • 5' a 85°C
The Real time reaction was carried out with TaqMan reagents (20x microRNA assay,
TaqMan noAmpErase UNG MasterMix-Applied Biosystems). All values were
usually normalized to endogenous control U6 snoRNA. miRNA quantification was
expressed in arbitrary units. For exosomes samples the normalization, instead, was
made on the basis of the starting number of cells and the medium volume of
exosomes preparation. We used the AB7900 instrument (Applied Biosystems).
Results
1. Characterization of Lung cancer stem cells (LCSCs)
Characterization of LCSCs is an area of active research and critical for developing
novel anti tumor therapies. The presence of a clonogenic population of cells in
human lung cancer was first described more than 25 years ago. Clinical specimens
from both adenocarcinoma and small-cell lung cancer (SCLC) patients contained
small populations of cells (<1,5%) that were able to form colonies in a soft agar
cloning assay. When individually selected soft agar colonies were injected into the
brain into athymic nude mice, they were able to generate tumors with features
characteristic of the original patient specimen, thus supporting the existence of a
CSC population in these malignancies (49. C.D. Peacock and D.N. Watkins, 2008).
Four main categories of lung tumors contribute to the vast majority of cases in terms
of both incidence and lethality. Small cell lung cancer (SCLC) is a neuroendocrine
tumor that represents about 20% of all lung cancers, while the most common forms
of the so-called non-SCLC (NSCLC) include adenocarcinoma (AC), squamous cell
carcinoma (SCC) and large cell carcinoma (LCC). Recent data indicates that stem
cells situated throughout the airways may initiate cancer formation. These putative
stem cells maintain pro-tumorigenic characteristics including high proliferative
capacity, multipotent differentiation, drug resistance and long lifespan when
compared to differentiated cells (50. Pine et al., 2008). The loss of differentiation
pathways could participate in maintenance of cancer stem cells.
We worked with different lung cancer stem lines (e.g. LCSC18, LCSC34, LCSC136)
obtained from Dott. Adriana Eramo. For the cultivation of the LCSC clones we used
flasks, which were not treated for tissue culture to reduce cell adherence and support
the growth as undifferentiated tumor spheres (Figure 10).
In a recent paper (51. Eramo et al., 2008) investigators have examined cancer stem
cells in the condition of lung cancer. The investigators found both in small cell and
non-small cell types of lung cancer that CD133 expression is associated with cells
containing "stem cell" properties.
Figure 10. LCSCs in culture. Lung cancer stem cells can be isolated from the major lung tumors
and then they can be expanded in vitro as tumor spheres.
The serum free stem cell medium was supplemented with growth factors (bFGF and
EGF) to ensure the maintenance of the cells as floating aggregates. Cultures were
expanded by mechanical dissociation of spheres, followed by re-plating of both
single cells and residual small aggregates in complete fresh medium. The stemness of
LCSCs was tested by growing them in a different culture medium, which induces
their differentiation. Figure 11 shows LCSC18 in two different states of
differentiation. In presence of increasing FBS concentrations the lung cancer stem
cells change their appearance, becoming adherent and acquire the morphology of
epithelial cells.
Figure 11. LCSCs differentiation. On the left: undifferentiated LCSCs. In the middle: LCSCs in
presence of DMEM 10% FBS for 24h; the cells still maintain their typical spherical morphology. On
the right: after 5 days, LCSCs in presence of FBS change their morphology.
In order to demonstrate that the lung cancer spheres indeed contain CD133+ cells, we
performed IF analysis on LCSC18, under stem cell conditions and the differentiated
counterpart (Figure 12). In the microscopic picture shown in figure it is evident that
CD133 surface protein decreases upon differentiation of LCSCs.
Figure 12. IF analysis for CD133 marker in Lung differentiated cells and in LCSCs. This
analysis demonstrated that surface marker CD133 is present in LCSC18 spheres, but the surface
protein is almost absent in differentiated lung cancer cells. DNA is shown in blue.
Having verified the cellular system, we started to develop a protocol for the isolation
of LCSC derived exosomes in several steps: Large scale exosome isolation from
Melanoma Stem Cells (MSCs) is well established in literature, thus these cells were a
good model system to study how to isolate and characterize exosomes. In order to
obtain exosomes from different LCSCs, the isolation protocol was based on the
protocol used for MSCs, but some slight modifications were introduced. Moreover
the exosomes isolation protocol was improved introducing a filtration and a
concentration steps. The complete procedure of the isolation is described in more
detail in the Materials and Methods section.
2. Biochemical characterization of exosomes
In order to confirm the presence of exosomes in our preparations, western blot
analysis for exosomal marker proteins was performed. The CD81 and CD9
tetraspanins, as well as TSG101 (tumor susceptibility gene) and CD71 (transferrin
receptor) proteins, are reported to be highly enriched on exosomes. Figure 13 clearly
shows that LCSC-derived exosomes show an enrichment of CD71 and TSG101
proteins when compared to corresponding LCSC lysates. Moreover we were able to
characterize LCSC136-derived exosomes also for the presence of CD9 tetraspanin,
which was not expressed by the other cell lines investigated. In all our samples the
very common exosome marker CD81 was not detected, however we cannot rule out,
that the antibody used for detection works well in western blot analysis. Taken
together, we could confirm the presence of typical exosome markers in our
preparation, suggesting a successful isolation of these nanovesicles from the culture
Figure 13. Biochemical characterization of exosomes. TSG101 and CD71 proteins are functional
exosomes markers of LCSC–derived exosomes; tetraspanin CD9 moreover is marker of LCSC136
clone; CD133 is strongly enriched in the LCSC34, 18 and 36-derived exosomes. LCSC136-derived
exosomes instead don't contain CD133 protein.
Western blot analysis for exosomal marker proteins showed also another important
result. We found, that CD133 protein is strongly enriched in the LCSC18, 34 and 36-
derived exosomes by comparison with the LCSCs lysates (Figure 13). LCSC136-
derived exosomes don't contain CD133 protein as well as LCSC136 lysate; instead
all the other typical exosomal markers such as TSG101, CD9 and CD71 proteins are
present in LCSC–derived exosomes as visible bands. We also detected by FACS
analysis, that LCSC136 don't show surface expression of CD133 (data not shown).
CD133 is a surface protein often used to define cancer stem cells; the structure of
CD133 protein shows homologies with proteins involved in plasma membrane
fusion. Therefore this finding was a stimulus to investigate its potential function in
the exosomes-mediated communication between tumor and cells of tumor
microenvironment.
3. CD133 is present on the surface to exosomes
The Immunoprecipitation (IP) technique allowed us to verify the presence of CD133
on the surface of exosomal vesicles. We used cells overexpressing CD133 in order to
verify if our method was functional. In order to obtain a CD133 producing cell line,
we first performed a lentiviral infection of HEK293T cells to obtain a stably
transduced HEK293TGFP control cell line and a HEK293T Prom1GFP cell line.
FACS analysis showed a high percentage of GFP-positive cells, indicating that the
HEK293T cells were successfully transduced (data not shown). In order to perform
IP technique we harvested supernatants from HEK293T Prom1GFP cells and from
HEK293TGFP cells (as a control), after 3-5 days in culture. As pointed out before,
exosomes display a typical markers composition such as TSG101 and also Rab5b.
We loaded the same amount of both control (GFP SN) and CD133 (Prom1 SN)
supernatants, incubated with biotinylated CD133 antibody and a biotinylated IgG1
antibody as a control. Both these antibodies are biotinylated; therefore the
Streptavidin-coupled Dynabeads used in this experiment could bind to the biotin
ligand. We found that only the immunoprecipitation using anti CD133 of the SN of
CD133 overexpressing cells shows the typical exosomal markers TSG101 and
Rab5B (Figure 14). Prom1 supernatant sample immunoprecipitated with CD133
antibody moreover shows an expected enrichment of CD133 protein. We performed
the same immunoprecipitation using other cells overexpressing CD133,
LCSC136GFP and LCSC136Prom1GFP supernatants and obtained similar result.
These experiments demonstrate for the first time, that LCSC-derived exosomal
vesicles are enriched in CD133 protein, which is marker of stemness. In addition, the
strong accumulation of CD133 in the exosomal particles suggests a physiological
role of this protein in the biological function of exosomes.
Figure 14. Immunoprecipitation experiment. This figure shows the immunoprecipitations
performed with control supernatant (GFP SN) and CD133 containing supernatant (Prom1 SN), both
incubated with biotinylated IgG1 and CD133 antibodies. Only Prom1 SN sample (highlighted in red)
immunoprecipitated with CD133 antibody shows typical exosomal markers TSG101 and Rab5b,
when compared with the other samples. This sample also shows an expected enrichment of CD133
protein.
4. CD133 is transported through exosomes into HEK293T cells
In order to investigate a potential function of CD133 in the intercellular trafficking of
exosomal particles, we investigated the transfer of CD133 from expressing to non-
expressing cells. In order to study the cellular uptake and the intracellular trafficking
of exosomes we used HEK293T cells as recipient cells.
First we performed experiments to investigate the potential uptake of exosomes by
recipient cells negative for CD133. Therefore we incubated HEK293T cells with
CD133 positive LCSC18 supernatant. Before incubation, the LCSC18 supernatant
was either ultracentrifuged or concentrated with the intent to enrich the exosomal
vesicles. FACS analysis, shown in figure 15, demonstrates that HEK293T cells
acquired high positivity (>80%) for CD133 protein after incubation with isolated
membrane particles from LCSC18 supernatant. This experiment was performed in
Figure 15. Uptake experiment with LCSC18 SN. FACS analysis of HEK293T cells for CD133.
The cells were incubated with pretreated, CD133 enriched supernatant from LCSC18; A) LCSC18
supernatant was ultracentrifuged at 33.000rpm; the ultracentrifuged pellet was resuspended in DMEM
and added to the cells for 12h; B) LCSC18 supernatant was concentrated (starting from 40ml of
supernatant) and added to the cells for 12h. After incubation with both SNs, HEK293T cells have
clearly acquired CD133 protein on their surface.
A similar experiment was performed testing supernatants from HEK293TGFP and
HEK293TProm1GFP stable transduced cell lines. We incubated GFP positive
recipient HEK293T cells with supernatants from HEK293TGFP (as a control) and
CD133 expressing HEK293TProm1GFP. FACS analysis in figure 16 shows that
HEK293T cells treated with control supernatant are GFP positive, but don't show
any positivity for CD133, instead the cells incubated with HEK293TProm1GFP
supernatant show a clear positivity (53%) for the stem cell marker. In this experiment
both supernatants were not concentrated. Thus, using the supernatant of
HEK293TProm1GFP cells as source of exosomal CD133 reduced the complexity of
the experiment, as ultracentrifugation or ultrafiltration steps could be omitted. The
experiment shown in figure 17 was performed in triplicate.
Figure 16. Uptake experiment with HEK293Prom1GFP SN. FACS analysis of HEK293T cells for
CD133. The cells were incubated with supernatants from HEK293TGFP (as control cell line) and
HEK293TProm1GFP (as CD133 cell line). A) HEK293T cells incubated for 16h with control SN
don't show any CD133 positivity; B) instead HEK293T cells incubated for 16h with Prom1
supernatant have acquired CD133 protein.
These experiments clearly demonstrated an uptake of CD133 by HEK293T cells. In
order to investigate the uptake of CD133 containing particles in more detail, we
performed time course experiments. HEK293T cells were incubated with
concentrated supernatant containing CD133 positive exosomes for different
timeframes. After incubation HEK293T cells were stained using a specific CD133
antibody for immunofluorescence and flow cytometric analysis. Figure 17 shows the
fluorescence microscopy pictures of HEK293T cells stained for CD133 (in red), for
Phalloidin which localizes on the cellular membrane (in green) and for DNA (in
blue); the CD133 protein was detected gradually around and inside the 293T cells.
After 30 minutes, more exosomes were internalized by cells and some exosomes
appeared at perinuclear regions. From 30 minutes to 240 minutes, CD133 containing
particles accumulated near nucleus and brighter and larger spots emerged. At early
time point of incubation we expected in cells a weak fluorescent intensity; instead
CD133 signal is kept bright until 240' time point.
We analyzed the CD133 uptake also by FACS analysis (Figure 18). After incubation
with concentrated and filtered CD133 supernatant, HEK293T cells were stained for
CD133. Non treated HEK293T cells are completely negative (0,3%) for CD133;
already after 30 minutes of treatment with CD133 supernatant, 96% of the cells
acquired the protein. After 60 minutes, the percentage of HEK293T cells that have
acquired CD133 protein is still high (89%); after 120 minutes to 240 minutes the
signal gradually decreases. Performing all these uptake experiments, we were able to
demonstrate that HEK293T cells negative for CD133, when incubated with CD133
enriched supernatant, acquired the stem cell protein; this uptake by HEK293T cells
can be due to the exosomal transport of CD133 protein.
Figure 17. Uptake of exosomes by HEK293T cells by IF. Immunofluorescence of HEK293T cells
for CD133 (shown in red, Phalloidin in green, DNA in blue). The cells were incubated at different
time points (30', 60', 120', 240') with CD133 enriched supernatant. After incubation HEK293T cells
have acquired CD133 protein. The negative control in the upper line doesn't show any CD133 signal.
Figure 18. Uptake of exosomes by HEK293T cells by FACS. FACS analysis of HEK293T cells for
CD133. The recipient cells were incubated at different time points with supernatant from HEK293T
Prom1GFP cells. Before incubation the supernatant was concentrated and filtrated.
5. Uptake Kinetic of exosomes by HEK293T cells
To investigate how long CD133 protein is detectable on and in HEK293T recipient
cells, we performed uptake kinetic experiments. We incubated HEK293T cells over
night with concentrated supernatants obtained from HEK293TGFP (control SN) and
HEK293TProm1GFP (CD133 SN) cells. The following day we removed the
supernatants and washed the cells and incubated them under normal culture
conditions. Different time points after the washing we stained the HEK293T cells for
CD133. The panel of immunofluorescences in figure 19 shows HEK293T cells
incubated over night with both supernatants; HEK293T cells were stained for CD133
exactly 15, 30, 60 and 120 minutes from the removal of the supernatants. We
detected CD133 around and inside the HEK293T cells treated with
HEK293TProm1GFP supernatant; 15 minutes after the removal of the supernatants,
HEK293T cells show a positive signal for CD133 (stained in red); going through the
other time points CD133 signal is still strong. At 60 and 120 minutes CD133,
accumulated inside the cells and also localized near nuclear region. The uptake of
CD133 protein is not detectable in all time points of recipient HEK293T cells treated
with control supernatant. Thus, CD133 is detectable in the HEK293T cells starting
from 15 up to 120 minutes after removal of the HEK293TProm1GFP supernatant.
We analyzed the uptake kinetic also by FACS analysis (Figure 20). HEK293T
recipient cells were incubated with either GFP or CD133 concentrated supernatants.
In order to investigate, how long CD133 remain on the surface of the recipient
HEK293T cells, we incubated the cells in SNs containing CD133 positive and
control exosomes. After 24 hours of incubation, we removed the SNs and washed the
cells. After the wash, we further cultivated the cells under normal cell culture
conditions and stained the HEK293T cells for CD133 at different time points after
removal of the SNs.
We found, that recipient cells keep CD133 signal until 2h, showing approximately
60% positivity. The results obtained by immunofluorescence and FACS analysis
indicated, that CD133-carrying exosomes were trapped in recipient cells, they
accumulated inside the cells and were transported actively to perinuclear region.
Then we investigated if target cells can be reactive to SN of CD133 positive cells.
Figure 19. Uptake Kinetic of CD133 positive exosomes by HEK293T cells investigated by IF.
Immunofluorescence of HEK293T cells for CD133 (shown in red, Phalloidin in grey, DNA in blue).
The cells were incubated over night with GFP control supernatant and with CD133 enriched
supernatant. The following day the cells were washed and stained for CD133at different time points.
HEK293T cells show CD133 protein until 120' after removal of supernatants. The HEK293T cells
incubated with control SN don't show CD133 signal never.
Figure 20. Uptake kinetic of CD133 positive exosomes by HEK293T cells investigated by FACS.
FACS analysis of HEK293T cells for CD133. The cells were incubated over night with supernatants
from HEK293T cells transfected with either ptwGFP or ptwProm1GFP vectors. HEK293T cells
treated with Prom1 enriched supernatant, at all time points, have acquired CD133 protein, showing a
clear positivity. Supernatant producing HEK293T cells were transfected with FuGENE reagent
(Roche).
6. CD133 can activate intracellular pathways
In order to investigate a potential role of CD133 in activation of cellular pathways in
cells incubated with CD133 containing exosomes, we performed a
phosphoproteomic-analysis of NIH3T3 cells. We treated these cells with SN of
CD133 expressing and non-expressing cells. After the incubation, we evaluated the
activity of MAP kinases. MAP kinases are involved in the development of many
tumors. We maintained NIH3T3 cells in DMEM medium without serum over night.
The following day we changed the medium of the recipient cells with either GFP
control or Prom1 GFP supernatants obtained from HEK293GFP and
HEK293TProm1GFP cell lines; both supernatants were obtained under serum free
conditions After different time points of incubation with CD133 enriched SN, we
collected and lysed the NIH3T3 cells in order to perform a western blot analysis of
cellular phospho-proteins. We investigated the following proteins: phospho-ERK,
phospho-p38, phospho-AKT, phospho-GSK3β, ERK1, AKT1. Western blot analysis
of these important kinases is shown in Figure 21. The western blot panel indicates a
significant increase in activation of phospho-ERK protein in NIH3T3 cells incubated
with Prom1 SN already 5 minutes after stimulation. Also at the 15 and 30 minutes
time points this activation is still evident. In addition, phospho-p38 shows a
significant increase in cells treated with Prom1 SN, especially 5 and 15 minutes after
the stimulation. The signal decreases at later time points (30 and 60 minutes).
Phospho-AKT has a strong activation 15 minutes upon treatment with Prom1 SN.
Instead phospho-GSK3β doesn't appears to increase in activation when treated with
Prom1 SN samples in comparison with the control. We used as control markers the
non-phosphorylated protein forms ERK1 and AKT1 and beta-actin as loading
control. These results highlight a potential role of CD133 protein in the activation of
MAP kinase pathway in cells having contact with CD133 containing exosomal
particles. In these cells especially the activation of ERK, p38 and AKT is
significantly increased, when compared to cells incubated with SNs of CD133
negative cells. In order to investigate if the intracellular activation is due to the
presence of CD133 on the surface of exosomes or is dependent on soluble factors
secreted by cells overexpressing CD133, we investigated a potential exosome-
mediated transfer of biological material from cell to cell.
Figure 21. Phosphoprotein analysis of NIH3T3 cells. Western blot (WB) analysis for several MAP
kinases after treatment with either GFP SN or Prom1 SN at different time points. A) Activation of
ERK at 5' and 15' time points of Prom1 SN samples; B) Activation of p38 at 5' and 15' time points of
Prom1 SN samples; C) Strong activation of Akt at 15' time point of Prom1 SN sample; D) Activation
of GSK3b; E) control markers.
7. Tetracycline-dependent regulatory system
The tetracycline-controlled transactivation system is one of the most powerful tools
for inducible expression of exogenous genes. This system enables selective, stringent
and quantitative regulation of transgenes in vitro and in vivo. The Tet-ON system
offers many advantages over other inducible gene expression systems. The inducer
has been used as an antibiotic for decades and it has been well characterized in
clinical settings. It is nontoxic at doses required for gene activation in preclinical and
clinical studies, and the margin of safety is high. Tetracycline (and its analogue
doxycycline) is rapidly metabolized and cleared from the body, making it an ideal
drug for the rapid increase in expression, long-term expression, and rapid decrease in
expression of the desired transgene. The components of the Tet-ON system
recognize unique sequences of DNA, and doxycycline does not interfere with native
proteins, reducing the potential of serious side effects when using this system.
However, because these proteins were derived from bacteria, they may be
immunogenic. These sophisticated methods are useful for investigation of gene
function during embryogenesis and in adult organs and for well-controlled gene
therapy approaches. The absence of doxycycline (Doxy) results in the inability of the
synthetic (Tet-activator) (tTA) to bind to the Tet operator (tetO) elements on the
reporter gene construct, therefore there is no promoter activation and subsequent
gene expression. The presence of Doxy results in its binding to the tTA, and then the
tTA/Doxy complex binds to the tetO sequences, allowing promoter activation and
gene expression (Figure 22).
Figure 22. Tetracycline-dependent regulatory system. The presence of Doxyclycline (Doxy)
results in its binding to the Transactivator tTA, and then the tTA/Doxy complex binds to the specific
tetO sequences, allowing promoter activation and gene expression (Luc gene); through this system the
luciferase activity can be measured.
8. CD133 mediates the transfer of biological materials from cell to cell
Considering that CD133 can be transferred through exosomes from expressing to
non-expressing cells, we set up an experimental system in order to detect the
functional transfer of proteins or genetic material from cell to cell. To perform this
transport experiment we selected two cell lines: CD133-positive HCT116 and
CD133-negative A549 cell lines (Figure 23). Chosen cell lines were permanent
transduced using two kinds of viral vectors, either TetO-CMV-Luc-GFP or rtTA3-
GFP. In order to perform a co-culture experiment the cell lines obtained,
HCT116Teto and HCT116rtTA3 were seeded in the same 12-well plate.
Figure 23. CD133 expression in HCT116 and A549 cell lines. A) FACS analysis of A549 and
HCT116 cells for CD133; B) Western blot analysis of A549 and HCT116 cells for CD133 protein. In
both analyses HCT116 cells are positive for CD133 protein, instead A549 cells are always negative.
We hypothesized that HCT116 rtTA3 cells could release rtTA3 activator molecule in
exosome like particles into the supernatants. These exosomes are consequently taken
up by TetO-CMV-Luc-GFP positive recipient cells and induce the activation of TetO
sequences and gene expression in presence of doxycycline (Figure 24).
Figure 24. Hypothesis of cell-to-cell transport of genetic material. Donor cell releases
transactivator molecules enclosed in vesicles. These vesicles can recognize the recipient cell through
receptor recognition and release their contents into the recipient cell. In this way the transactivator
molecule can bind the TetO sequences and in presence of doxycycline activate luciferase gene
expression.
To verify our experimental system, we performed a control experiment. We
transfected either HCT116 control cells or stably transduced HCT116rtTA3 cells
with the Teto-Luc construct. Then, we verified the luciferase activity in presence of
doxyclycline. In presence of doxycycline HCT116 control cells transfected with the
TetO-Luc construct don't show any induction in luciferase activity. In contrast
HCT116 rtTA3 cells transfected with the same construct show a strong induction of
luciferase activity in presence of doxycycline (Figure 25).
Figure 25. HCT116 control experiment. Our reporter system is functional; in presence of
doxyclycline we observe a strong luciferase activity in HCT116 rtTA3 stably transduced cells which
are transfcted with TetO construct; we can't observe any induction when doxycycline is absent.
We then performed co-culture experiments seeding in the same well either
HCT116TetO cells with HCT116 control cells or HCT116TetO cells with
HCT116rtTA3 cells in presence or absence of doxycycline. We performed this
experiment seeding two different amounts of HCT116 rtTA3 cells, three times and
nine times more than HCT116 TetO cells. In both cases our results of the co-culture
experiments show an induction of luciferase activity in TetO-luc cells when co-
cultured with rtTA3 cells in presence of doxycycline. This effect could be increased
by the coexpression of a viral protein (vesicular stomatitis virus (VSV) protein G),
which is known to lead to cell fusion events and was added as positive control to the
experiment. In contrast, in the control samples we cannot detect any induction of
luciferase activity (Figure 26). These results suggest an interchange of genetic
information or protein between both stably transduced cell lines.
Figure 26. Co-Culture experiment with HCT116 and A549 cell lines. A) GFP FACS analysis of
CD133 positive HCT116 cells in order to control transduction efficiency; B) GFP FACS analysis of
CD133 negative A549 cells in order to control stable transduction efficiency; C) A549rTA3 cells in
co-culture with A549TetO cells whether in presence or in absence of doxycycline don't show any
luciferase induction; HCT116rtTA3 cells in co-culture with HCT116TetO cells in presence of
doxycycline show a good induction of luciferase activity; HCT116rtTA3 cells in co-culture with
HCT116TetO cells in presence of doxycycline and of pMD2 vector show a bigger luciferase activity
than HCT116TetO + HCT116rtTA3 co-culture.
9. Secretory exosomes are active vehicles for intercellular miRNA
transfer
After we got some indications, that CD133 supports an active transport of biological
material from cell to cell, we investigated a potential role of CD133 enriched
exosomes in the intercellular transfer of microRNAs (miRs). In order to verify if and
how much RNA we were able to extract from our exosomes preparation first we
extracted total RNA from supernatants of HEK293T control cells and stable
transduced HEK293TProm1GFP cells, obtaining: 1,9µg from HEK293T cells and
3,3µg from 293TProm1GFP.
In order to estimate the exosomes mediated transfer from cell to cell we selected
miR-146a on the basis of its quantitative miRNA expression in the recipient cells.
We first evaluated the miR-146a expression in the HEK293T cells when incubated
with LCSC136-derived exosomes. The histogram in the figure 27A shows the fold
induction of the miR-146a expression; HEK293T cells incubated with LCSC136-
derived exosomes have acquired miR146a about five times compared to the control
sample indicating that LCSC136-derived exosomes transport this miRNA and they
can transfer this genetic material in target cells. Then we incubated HEK293T
LCSC136Prom1GFP-derived exosomes to demonstrate that CD133 on the exosomes
surface is able to enhance mir-146a transfer. We were indeed able to extract RNA
from the same amounts of concentrated supernatants and we obtained 6,2µg of RNA
from LCSC136-derived exosomes and 4,6 µg of RNA from LCSC136Prom1GFP-
derived exosomes; the histogram in the figure 27B shows the three conditions of
miR-146a expression: HEK293T control sample, HEK293T cells incubated with
LCSC136-derived exosomes, which have acquired miR146a also in this experiment
about five times compared to the control sample and HEK293T cells incubated with
LCSC136Prom1GFP-derived exosomes, which have acquired miR146-a about eight
times compared to the control. We repeated this experiment three times. These
results suggest that, even by starting from a smaller amount of RNA contained in the
CD133 enriched exosomes, LCSC136Prom1GFP-derived exosomes seem to be able
to transfer a larger quantities of miR146-a than LCSC136-derived exosomes.
Therefore CD133 localized on exosomes vesicles could help the intercellular miRNA
Figure 27. Transfer through CD133 enriched exosomes of miR-146a. A) HEK293T recipient cells
incubated with LCSC136-derived exosomes acquire miR146a about five times compared to the
HEK293T control sample; B) HEK293T recipient cells incubated with LCSC136Prom1GFP-derived
exosomes acquire about eight times compared to the HEK293T control sample.
10. Immunoprecipitated CD133 positive exosomes contain miR-146a
In order to delve into this result and to verify if CD133 containing exosomes, indeed
transport microRNAs, we performed immunoprecipitation of LCSC136GFP and
LCSC136Prom1GFP supernatants using both IgG1 antibody control and CD133
Milteniy antibody. By immunoprecipitation we specifically extracted the exosomal
fraction from the supernatants. Using paramagnetic beads, we were able to
specifically pull down CD133 positive exosomal vesicles. We lysed the
immunoprecipitation products and extracted their microRNA content. The histogram
in figure 28 shows the expression of the miR-146a in the immunoprecipitated
exosomes. We detected a strong accumulation of miR-146a attached to the beads
used for the pull-down of CD133, when compared to the control beads. This
experiment was repeated three times. These results revealed that we didn't isolate
just protein aggregates, but CD133 positive exosomes, which are able to transport
microRNAs. This experiment confirms the presence of microRNAs in the exosomal
vesicles demonstrating that exosomes can have a role in the communication between
the cells and above all that CD133 enriched exosomes are involved in the miRNAs
transfer between the cells.
Figure 28. Immunoprecipitation of CD133 enriched exosomes containing miR146a. LCSC136-
derived exosomes and LCSC136Prom1-derived exosomes were obtained by immunoprecipitation
method; both kinds of exosomes were lysed and the microRNAs content was extracted. We used for
this experiment the 15% of each IP. Exosomes contain miR146a. The experiment was repeated three
times.
Discussion
In order to understand tumor biology in more detail, we investigated one aspect of
the interactions between tumor and host cells in the tumor microenvironment. Our
hypothesis was that exosomal vesicles, released by tumor cells, are responsible
interchange of information in the tumor microenvironment. Exosomes are known to
be released from a big number of tumor cells and therefore thought to play an
important role in tumor biology. Whereas some basic principles of exosome
biogenesis and secretion and their influence on the immune system (52. Keller et al.,
2009) are known, important aspects such as a potential pro-tumorigenic role and
mode of action remain still unclear. Here we analyzed exosomes secreted by LCSCs.
We were able to determine an ideal procedure to isolate high amounts of exosomes
from LCSC supernatants. We confirmed our isolation protocol by detecting typical
exosomes markers, such as TSG101, CD71 and CD9 in our exosome preparations.
Going on with the characterization of the exosomes, we found that LCSCs-derived
exosomes contain high amounts of CD133 protein. By immunoprecipitation
technique we demonstrated the mutual correlation between exosomal markers and
CD133 presence in the same particles, which is in contradiction to reports of
Marzesco et al., which describes two different types of particles either containing
exosomal proteins or CD133. In this work Marzesco et al. analyzed the "prominin-1-
containing P4 particles". They asserted that these particles, though similar in size to
exosomes, in fact represented a subpopulation of small membrane vesicles distinct
from exosomes, lacking immunoreactivity for CD63, a tetraspan membrane protein
highly enriched in exosomes from virtually any cell type (53. Marzesco et al., 2005).
This contradiction might be due to the different model systems used in the two
studies or the detection methodology. While we used either the CD133 containing
particles from lung cancer stem cells or HEK293T overexpressing CD133, Marzesco
et al. used a colon carcinoma cell line (Caco-2). In addition, we biochemically
purified CD133-containing particles by immunoprecipitaion and analyzed the
purified proteins by western blot, Marzesco et al. detected exosomal proteins versus
CD133 by electron microcopy (54. Marzesco et al., 2005).
The strong accumulation of CD133 in the exosomal particles suggested a
physiological role of this protein in the biological function of exosomes.
Increasing evidence highlights the function of CD133 as a marker of CSCs in various
human tumors. Previously, it was described that CD133-expressing cells in brain
tumors have the capacity for unlimited self-renewal, as well as the ability to initiate
tumor formation and progression in immuno-deficient mice, when injected at low
numbers (55. Singh et al., 2004), suggesting that CD133-expressing cells fulfill
important criteria required for tumor-initiating cells (56. Jordan et al., 2006);
however, its function in tumorigenesis remains to be elucidated.
Although the interaction between exosomes and cells has been verified and many
important functions of exosomes are being revealed, the exact mechanism of how
exosomes enter cells and their trafficking in cells are still unclear. Due to its large
size, endocytosis and fusion can be the two possible mechanism of exosomes uptake
(57. Morelli et al., 2004).
In this study we focused our attention on the release of CD133 containing exosome-
like particles from the CD133-expressing LCSC lines and conventional cell lines
ectopically overexpressing CD133. We investigated a potential role of CD133 in the
transfer of these vesicles from expressing to non-expressing cells and in the transport
of genetic and biological material, such as microRNAs, from cell to cell.
The concept that cells can directly communicate with and influence the function of
other cells by transferring particular complexes or cell surface proteins rather than
soluble factors has excited cell biologist for decades (58. Ling-Pei Ho, 2010).
Extensive efforts have been made to prove the existence of this phenomenon and
understand the mechanism by which cells transfer proteins between each other.
Recent studies demonstrated that exosomes are a significant example of protein
transfer between cells (59. Ling-Pei Ho, 2010).
We performed uptake experiments by immunofluorescence and FACS analysis. Our
exosome uptake experiments, using HEK293T as recipient cells, demonstrated that
CD133 is transferred through exosomes on recipient cells.
We demonstrated that HEK293T cells negative for CD133, when incubated with
CD133 enriched supernatant, acquired the stem cell protein; this uptake by
HEK293T cells can be due to the exosomal transport of CD133 protein.
This allowed us to assert that exosomes are functional vesicles, which are able to
transfer protein material.
By uptake kinetic experiments we found that recipient cells keep the signal for
CD133 until 2h, showing approximately 60% positivity. We were also able to detect
a perinuclear accumulation of CD133 upon the uptake of the vesicles from the
extracellular space. This specific intracellular trafficking together with the fact, that
the CD133 was not readily degraded, let us hypothesize, that we do not detect the
passive endocytosis of the CD133 positive exosomes, but a coordinated process. For
this reason we went on and investigated a potential role of CD133 in activation of
cellular pathways in cells treated with the SNs of CD133 expressing cells. Treating
recipient cells with SN enriched with CD133 containing particles, we found that
CD133 is able to activate intracellular pathways, connected with enhanced
proliferation and apoptosis resistance in target cells. We indeed demonstrated that
recipient cells (NIH3T3) incubated with CD133 enriched supernatants show the
activation of MAP kinases pathway, in particular phospho-ERK, phospho-p38 and
phospho-AKT proteins are increased. This pathway is a frequent event in
tumorigenesis; MAPKs have been implicated in cell migration, proteinase induction,
regulation of apoptosis and angiogenesis, events that are essential for successful
completion of metastasis (60. Reddy et al., 2003). Therefore this result suggests a
potential role of CD133 in the tumor progression through MAP kinases activation.
Interestingly, a recent study in glioblastoma has shown that extracellular vesicles
transport RNA and proteins that promote tumor growth and provide diagnostic
biomarkers (61. Skog et al., 2008). Using a tetracycline-dependent regulatory system
in the stable transduced cell lines HCT116rtTA3 and HCT116TetO, we observed that
CD133 containing exosome-like particles could transfer biologically active
intracellular proteins or mRNA from cell to cell.
HCT116rtTA3 donor cell line released transactivator molecules enclosed in vesicles,
which recognized the HCT116TetO recipient cell through receptor recognition
releasing their contents inside. In this way the transactivator molecule can bind to the
TetO sequences and activated luciferase gene expression in presence of doxycycline.
Using this elegant model system, we verified an interchange of genetic information
or protein between two stably transduced cell lines, without the need of transient
transfection and all the potential artifacts connected to this method.
MicroRNA-expression profiling can be used as diagnostic tool for cancers that
currently lack reliable molecular markers and recent studies have reported the
association of microRNA with circulating tumor-derived exosomes (62. Taylor and
Exosomes have been postulated to play an important role in cell-cell communication
and appear to affect target cells either by stimulating them directly by surface
expressed ligands or by transferring molecules between cells (63. Taylor and Taylor,
Taylor and Taylor demonstrated that microRNA contained in tumor exosomes is
functional and can suppress the mRNA for signal transduction components within T
cells. Ratajczak et al. (64) demonstrated the presence of exosomal RNA and
provided evidence for the horizontal transfer of genetic information between cells.
The biological effects of these exosomes were inhibited after pretreatment with
RNAse, indicating the involvement of RNA components. The results indicate that
RNA molecules, following translocation from the nucleus to the cytoplasm, can bind
to and are transported by membranous organelles or vesicles to specific intracellular
sites. This is a potential explanation for the association of RNAs with exosomes.
Valadi et al. (65) demonstrated that exosomes carry both mRNA and miRNA when
released from mast cells and that many of these RNA species seem to be specifically
packaged exclusively into exosomes. They also confirmed that the mRNA in their
exosomes is transfectable. Interestingly, the proteins encoded by these transported
mRNAs were often exosome specific (66. Valadi et al., 2007). All these report how
evidence, that exosomes contain a subset of both cellular mRNA and microRNA,
which could be transferred to target cells.
As also described in previous studies, also we could show, that miR146-a contained
in LCSC-derived exosomes can be transferred to HEK293T recipient cells. In
addition we found, that CD133 containing exosome-like particles from the CD133-
expressing LCSC lines are able to transfer larger amounts of miR146-a to HEK293T
cells than LCSC-derived exosomes not containing CD133. These findings point
towards a new, highly interesting function of CD133, which was studied intensively
in the last decade due to its restricted expression pattern, mainly on stem and cancer
stem cells. Our findings might help to elucidate the interplay between cancer stem
cells and the cells in the tumor microenvironment, which have important functions
also in regulating the "stemness" of the CSCs.
The enhanced uptake of CD133 containing exosomes, containing functional
biological material such as miRNAs and most probably also a number of other
proteins, by microenvironmental cells, might be a way, of how CD133 positive CSCs
can reprogram these cells to their advantage. We have now to directly correlate the
changes in the MAP and AKT pathways of recipient cells treated with CD133
containing particles and try to find out, which factors are indeed responsible for our
Cell-cell communication can occur by several means, including chemical receptor-
mediated events, direct cell-cell contact and cell-cell synapse (67. Valadi et al.,
2007). Here, we suggest that another mechanism of cell-cell communication can be
delivery of RNA by transfer through exosomes, increasing the complexity by which
cells may communicate. In summary LCSC-derived exosomes are active functional
vesicles and the presence of CD133 protein on their surface could be a decisive role
in this transport, increasing its efficiency. This protein and genetic communication
between cells may be occurring in the microenvironment, but could also occur at a
distance, by traffic of exosomes. We contributed to elucidate a potential role of
CD133 protein, which until now is the official marker of stemness in many solid
tumors, but its real function should be delved into more detail.
I acknowledge the invaluable guide of Dr. Tobias Haas, who followed me up and
sustained during the PhD training; I also want acknowledge the countless advices of
Dr. Mauro Biffoni and his precious FACS analysis. I acknowledge the Prof. Daniele
Condorelli and Prof. Vincenza Barresi for their helpfulness and suggestions. Finally
I acknowledge the most important Prof. Ruggero De Maria, who gave to me the
opportunity to conduct in his department my PhD project.
1. The organizing principle: microenvironmental influences in the normal and
malignant breast. Differentiation. 2002 Dec; 70 (9-10): 537-46 Bissell MJ,
Radisky DC, Rizki A, Weaver VM, Petersen OW.
2. The hallmarks of cancer. Cell. 2000 Jan 7; 100(1): 57-70. Hanahan D, Weinberg
3. Inflammation and cancer. Nature. 2002 Dec 19-26; 420(6917): 860-7. Coussens
4. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer. 2008
Nov 15;123 (10):2229-38. De Wever O, Demetter P, Mareel M, Bracke M.
5. Angiogenesis in cancer and other diseases. Nature. 2000 Sep 14;407 (6801):249-
57. Carmeliet P, Jain RK.
6. Tumor angiogenesis. N Engl J Med. 2008 May 8 ;358(19):2039-49. Kerbel RS.
7. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003 Jun;3 (6):401-
10. Bergers G, Benjamin LE.
8. Reference number 3.
9. Reference number 6.
10. The tumour microenvironment as a target for chemoprevention. Albini A, Sporn
MB. Nat Rev Cancer. 2007 Feb;7 (2):139-47.
11. G. van Niel, I. Porto-Carreiro, S. Simoes, G.Raposo/ J Biochem.( 2006)
12. S. Keller S et al. Biochem J. 2006 Feb 1;393(Pt 3):609-18
13. Applying the principles of stem-cell biology to cancer. Pardal R, Clarke MF,
Morrison SJ. Nat Rev Cancer. 2003 Dec;3 (12):895-902.
14. The biology of cancer stem cells. Lobo NA, Shimono Y, Qian D, Clarke MF.
Annu Rev Cell Dev Biol. 2007;23:675-99.
15. Prospective identification of tumorigenic breast cancer cells. Al-Hajj M, Wicha
MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Proc Natl Acad Sci U S A.
2003 Apr 1;100 (7):3983-8.
16. Identification of a cancer stem cell in human brain tumors. Singh SK, Clarke ID,
Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Cancer Res. 2003 Sep
15;63 (18):5821-8.
17. Prospective identification of tumorigenic prostate cancer stem cells. Collins AT,
Berry PA, Hyde C, Stower MJ, Maitland NJ. Cancer Res. 2005 Dec 1;65
18. Stem cells and lung cancer: future therapeutic targets? Malcolm R Alison,
Arielle C Le Brenne & Shahriar Islam. Expert Opin. Biol. Ther. (2009)
19. Reference number 18.
20. Cancer stem cells and the ontogeny of lung cancer. Peacock CD, Watkins DN. J
Clin Oncol. 2008 Jun 10;26 (17):2883-9.
21. Reference number 20.
22. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell
differentiation. Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L,
Hoek M, Zeilstra J, Pals ST, Mehmet H, Stassi G, Medema JP. Cancer Res. 2010
Jan 15 ;70(2):719-29.
23. CD133: molecule of the moment. J Pathol. 2008 Jan;214 (1):3-9. Mizrak D,
Brittan M, Alison MR.
24. Detection of tumor stem cell markers in pancreatic carcinoma cell lines.
Hepatobiliary Pancreat Dis Int. 2007 Feb;6(1):92-7. Olempska M, Eisenach PA,
Ammerpohl O, Ungefroren H, Fandrich F, Kalthoff H.
25. CD133 as a marker for cancer stem cells: progresses and concerns. Wu Y, Wu
PY. Stem Cells Dev. 2009 Oct;18(8):1127-34.
26. Prominin-1 (CD133) Is Not Restricted to Stem Cells Located in the Basal
Compartment of Murine and Human Prostate. The Prostate. Ewa Missol-Kolka,
Jana Karbanova, Peggy Janich, Michael Haase, Christine A. Fargeas, Wieland B.
Huttner, and Denis Corbeil.
27. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell
differentiation. Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L,
Hoek M, Zeilstra J, Pals ST, Mehmet H, Stassi G, Medema JP. Cancer Res. 2010
Jan 15 ;70(2):719-29.
28. CD133: molecule of the moment. J Pathol. 2008 Jan;214 (1):3-9. Mizrak D,
Brittan M, Alison MR.
29. The intriguing links between prominin-1 (CD133), cholesterol-based membrane
microdomains, remodeling of apical plasma membrane protrusions, extracellular
membrane particles, and (neuro)epithelial cell differentiation. Denis Corbeil a,
Anne-Marie Marzesco b, Michaela Wilsch-Bräuninger b, Wieland B. Huttner.
FEBS Letters 584 (2010) 1659–1664.
30. CD133: molecule of the moment. J Pathol. 2008 Jan;214 (1):3-9. Mizrak D,
Brittan M, Alison MR.
31. Exosomes: A Common Pathway for a Specialized Function. Guillaume van Niel,
Isabel Porto-Carreiro, Sabrina Simoes and Gra¸ca Raposo. J. Biochem. 140, 13–
32. Reference number 31.
33. Reference number 31.
34. Exosomes: from biogenesis and secretion to biological function. Keller S,
Sanderson MP, Stoeck A, Altevogt P. Immunol Lett. 2006 Nov 15;107(2):102-8.
Epub 2006 Oct 17.
35. Exosomes/microvesicles: mediators of cancer-associated immunosuppressive
microenvironments. Douglas D. Taylor and Cicek Gercel-Taylor. Seminars in
Immunopathology Volume 33, Number 5, 441-454.
36. MHC Class II+ Exosomes in Plasma Suppress Inflammation in an Antigen-
Specific and Fas Ligand/Fas-Dependent Manner. Adrian E. Morelli and Paul D.
Robbins Seon Hee Kim, Nicole R. Bianco, William J. Shufesky. J Immunol
2007;179;2235-2241
37. Exosome function: from tumor immunology to pathogen biology. Schorey JS,
Bhatnagar S. Traffic. 2008 Jun;9 (6):871-81. Epub 2008 Mar 6.
38. Exosomes: secreted vesicles and intercellular communications. C. Thery.
F1000 Biol Reports 2011, 3:15.
39. Tumor initiating cells in malignant gliomas: biology and implications for
therapy. Costas G. Hadjipanayis and Erwin G. Van Meir. Journal of Molecular
Medicine Volume 87, Number 4, 363-374.
40. MicroRNAs: key regulators of stem cells. Nat Rev Mol Cell Biol. 2009
Feb;10(2):116-25. Gangaraju VK, Lin H.
41. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Hatley
ME, Patrick DM, Garcia MR, Richardson JA, Bassel-Duby R, van Rooij E,
Olson EN. Cancer Cell. 2010 Sep 14;18(3):282-93.
42. MicroRNA signatures in human cancers. Calin GA, Croce CM. Nat Rev Cancer.
2006 Nov;6 (11):857-66.
43. Modulation of K-Ras-Dependent Lung Tumorigenesis by MicroRNA-21. Mark E.
Hatley, David M. Patrick, Matthew R. Garcia, James A. Richardson, Rhonda
Bassel-Duby, Eva van Rooij and Eric N. Olson. Cancer Cell 18, 282–293,
September 14, 2010.
44. Reference number 43.
45. Secretory microRNAs as a versatile communication tool. Iguchi H, Kosaka N,
Ochiya T. Commun Integr Biol. 2010 Sep;3(5):478-81.
46. Multivesicular bodies associate with components of miRNA effector complexes
and modulate miRNA activity. Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O.
Nat Cell Biol. 2009 Sep;11(9):1143-9.
47. Secretory microRNAs as a versatile communication tool. Iguchi H, Kosaka N,
Ochiya T. Commun Integr Biol. 2010 Sep;3(5):478-81.
48. Secretory Mechanisms and Intercellular Transfer of MicroRNAs in Living Cells.
Nobuyoshi Kosaka, Haruhisa Iguchi, Yusuke Yoshioka, Fumitaka Takeshita, and
Takahiro Ochiya. J.Biol.Chem. 2010, 17442-52.
49. Cancer stem cells and the ontogeny of lung cancer. Peacock CD, Watkins DN. J
Clin Oncol. 2008 Jun 10;26 (17):2883-9.
50. Lung cancer stem cells. Sharon R. Pine, Blair Marshall and Lyuba Varticovski.
Disease Markers 24 (2008) 257-266.
51. Identification and expansion of the tumorigenic lung cancer stem cell population.
Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C,
Ruco L, Peschle C, De Maria R. Cell Death Differ. 2008 Mar;15(3):504-14
52. Systemic presence and tumor-growth promoting effect of ovarian carcinoma
released exosomes. Keller S, König AK, Marmé F, Runz S, Wolterink S,
Koensgen D, Mustea A, Sehouli J, Altevogt P. Cancer Lett. 2009 Jun
53. Release of extracellular membrane particles carrying the stem cell marker
prominin-1 (CD133) from neural progenitors and other epithelial cells.
Marzesco AM, Janich P, Wilsch-Bräuninger M, Dubreuil V, Langenfeld K,
Corbeil D, Huttner WB.) J Cell Sci. 2005 Jul 1; 118: 2849-58.
54. Reference number 53.
55. Identification of human brain tumour initiating cells. Singh SK, Hawkins C,
Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks
PB. Nature. 2004 Nov 18;432 (7015):396-401.
56. Cancer stem cells. Jordan CT, Guzman ML, Noble M. N Engl J Med. 2006 Sep
21;355(12):1253-61.
57. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells.
Blood. 2004 Nov 15;104 (10):3257-66. Morelli AE, Larregina AT, Shufesky WJ,
Sullivan ML, Stolz DB, Papworth GD, Zahorchak AF, Logar AJ, Wang Z,
Watkins SC, Falo LD Jr, Thomson AW.
58. Exosomes in lungs of patients with sarcoidosis: a contributor to immune
pathogenesis or just another by-product of heightened immune activity? Thorax.
2010 Nov; 65 (11): 947-8. Ling-Pei Ho.
59. Reference number 58.
60. Role of MAP kinase in tumor progression and invasion. Reddy KB, Nabha SM,
Atanaskova N. Kerbel. Intr Cancer Metastasis Rev. 2003 Dec;22(4):395-403.
61. Glioblastoma microvesicles transport RNA and proteins that promote tumour
growth and provide diagnostic biomarkers. Skog J, Würdinger T, van Rijn S,
Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky
AM, Breakefield XO. Nat Cell Biol. 2008 Dec;10(12):1470-6.
62. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of
ovarian cancer. Taylor DD, Gercel-Taylor C. Gynecologic Oncology 110 (2008)
63. Reference number 62.
64. Embryonic stem cell-derived microvesicles reprogram hematopoietic
progenitors: evidence for horizontal transfer of mRNA and protein delivery.
Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, Ratajczak MZ.
Leukemia. 2006 May;20 (5):847-56.
65. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of
genetic exchange between cells. Valadi H, Ekström K, Bossios A, Sjöstrand M,
Lee JJ, Lötvall JO. Nat Cell Biol. 2007 Jun;9(6):654-9.
66. Reference number 65.
67. Reference number 65.
Source: http://archivia.unict.it/bitstream/10761/921/1/BRNLDI81P67G273E-Lidia%20Brunetto's%20PhD%20THESIS_High_A.pdf
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use ------------------------WARNINGS AND PRECAUTIONS ------------------------ ZOSTAVAX safely and effectively. See full prescribing information Hypersensitivity reactions including anaphylaxis have occurred for ZOSTAVAX.
Annual Report for the year ending 31st December 2009 Investing in your FutureSupporting the Creation & Development of Local Enterprise Mike Cantwell Asst. CEO & Mairead Barron St. Joseph's Sean Gallagher, Ned Toomey CEO & Secondary School Doon, Co. Limerick Agnes Relihan Business Advisor New Horizons Networking Event with Mid