Lncc.br
Genetics and Molecular Biology, 27, 4, 605-610 (2004)
Copyright by the Brazilian Society of Genetics. Printed in Brazil
A genetic algorithm for the ligand-protein docking problem
Camila S. de Magalhães1, Hélio J.C. Barbosa1 and Laurent E. Dardenne2
1Laboratório Nacional de Computação Científica, Departamento de Matemática Aplicada
e Computacional, Petrópolis, RJ, Brazil.
2Laboratório Nacional de Computação Científica, Departamento de Mecânica Computacional,
Petrópolis, RJ, Brazil.
We analyzed the performance of a real coded "steady-state" genetic algorithm (SSGA) using a grid-basedmethodology in docking five HIV-1 protease-ligand complexes having known three-dimensional structures. Allligands tested are highly flexible, having more than 10 conformational degrees of freedom. The SSGA was tested forthe rigid and flexible ligand docking cases. The implemented genetic algorithm was able to dock successfully rigidand flexible ligand molecules, but with a decreasing performance when the number of ligand conformational degreesof freedom increased. The docked lowest-energy structures have root mean square deviation (RMSD) with respectto the corresponding experimental crystallographic structure ranging from 0.037 Å to 0.090 Å in the rigid docking, and0.420 Å to 1.943 Å in the flexible docking. We found that not only the number of ligand conformational degrees offreedom is an important aspect to the algorithm performance, but also that the more internal dihedral angles arecritical. Furthermore, our results showed that the initial population distribution can be relevant for the algorithmperformance.
Key words: ligand-protein docking, flexible docking, genetic algorithms.
Received: September 22, 2003; Accepted: May 12, 2004.
rigid body molecules considering only the ligand
With the increasing amount of molecular biological
translational and orientational degrees of freedom (Ewing
structures available, docking approaches have been very
and Kuntz, 1997). Other docking algorithms also include
important and useful tools in structure-based rational drug
the ligand flexibility and account for the ligand
discovery and design (Gane and Dean, 2000). For a pro-
conformational degrees of freedom (Jones
et al., 1997;
tein/receptor with known three-dimensional structure, the
Rarey
et al., 1996). In the two docking classes above, the
ligand-protein docking problem basically consists in pre-
protein structure is fixed in the position of the experimental
dicting the bound conformation of a ligand molecule within
crystallographic structure. Docking large, highly flexible
the protein active site. The docking problem is a difficult
ligands is still a challenge for even the most sophisticated
optimization problem involving many degrees of freedom,
current docking algorithms (Wang
et al., 1999), and adding
and the development of efficient docking algorithms and
the receptor flexibility remains a major challenge (Carlson
methodologies would be of enormous benefit in the design
and McCammon, 2000).
of new drugs (Marrone
et al., 1997). One of the major prob-
Genetic Algorithms are inspired in Darwin's theory
lems in molecular docking is how to treat the protein and
of evolution by natural selection and are powerful tools in
the ligand flexibility, taking into account hundreds of thou-
difficult search and optimization problems (Holland, 1975;
sands of degrees of freedom in the two molecules. In the
Goldberg, 1989).
last few years several docking programs have been devel-
They have been shown to be a promising search algo-
oped (Diller and Verlinde, 1999; McConkey
et al., 2002).
rithm for the ligand-protein docking problems (Morris
et
Some docking programs treat the receptor and the ligand as
al., 1998). The GA works with a population of individuals
where each individual represents a possible solution for the
Send correspondence to Camila Silva de Magalhães. Laboratório
problem to be solved and, in ligand-protein docking prob-
Nacional de Computação Científica, Departamento de Matemática
lem, it is the position of the ligand with respect to the pro-
Aplicada e Computacional, Av. Getúlio Vargas 333, sala 1A-24,25651-075 Quitandinha, Petrópolis, RJ, Brazil. E-mail: camilasm@
tein. Therefore, a ligand conformation is represented by a
chromosome constituted by real valued genes representing
Magalhães
et al.
ligand translational, orientational and conformational de-
conformational genes are the ligand dihedral angles (one
grees of freedom. The individuals are evaluated by a fitness
gene to each dihedral angle). The ligand-protein energy
function, that is, the total interaction energy between the
function used is the GROMOS96 (van Gunsteren and
protein and the ligand molecule and the intramolecular
Berendsen, 1987; Smith
et al., 1995) classical force field
ligand energy. Individuals in the population are selected for
implemented in the THOR (Pascutti
et al., 1999) program
reproduction in accordance with their fitness, and undergo
of molecular mechanics/dynamics. The force field parame-
mutation and crossover reproduction operators, to generate
ters are adjusted to reproduce experimental results (
e.g.,
new individuals. In this paper, a non-generational also re-
structural and thermodynamic properties) or higher level
ferred to as steady-state GA (Whitley, 1995) is adopted. In
ab initio quantum calculations (Brooks III
et al., 1988). The
a steady-state GA (SSGA) there is no separation between
GROMOS force field is given by:
consecutive generations of the population. Instead, each
offspring is created and immediately tested for insertion in
the population. In the following, the term generation will be
Protein Ligand rij
Drij LigandLigand rij
associated with the creation of a single offspring (candidate
∑ k(1+ cos(ϖ θkk −θ0k))
solution) and its evaluation. The variable maxgen will thus
denote the maximum number of objective function evalua-
tions (which is equal to the total number of offspring gener-
where rij is the distance between the atoms i and j; Aijand Bij
ated). A pseudo-code for the steady-state GA used here is
are the Lennard-Jones parameters; qi and qj are atomic
displayed as follows:
charges and D is a sigmoidal distance-dependent dielectric
constant (Arora and Jayaram, 1997).
Initialize the population
P
The first term of the equation corresponds to van der
Evaluate individuals in
P
Waals interaction and electrostatic interaction between the
Sort
P according to the fitness value
protein and the ligand molecule, and the last two terms cor-
respond to the ligand internal energy interaction, which
select genetic operator
also have one term for van der Waals interaction and one
select individual(s) for reproduction
term for electrostatic interaction. The ligand-protein dock-
apply genetic operator
ing problem involves millions of energy evaluations, and
evaluate offspring
the computational cost of each energy evaluation increases
select individual
xi to survive
with the number of the atoms of the complex ligand-protein
if
xi is better than worst individual in
P then
which has thousands of atoms. To reduce the computational
remove worst individual from
P
cost, we implemented a grid-based methodology where the
insert
xi in
P according to its rank
protein active site is embedded in a 3D rectangular grid and
on each point of the grid the electrostatic interaction energy
until stopping criteria are met
and the van der Waals terms for each ligand atom type are
pre-computed and stored, taking into account all the protein
The SSGA differs from traditional GA basically by
atoms. In this way the protein contribution at a given point
applying only one operator and replacing only one individ-
is obtained by tri-linear interpolation in each grid cell. A
ual in each generation. In this work, we are interested in
random initial population of individuals is generated inside
testing the use of a SSGA using a grid-based methodology
the grid. For translational genes, random values between
in the rigid and flexible ligand docking cases. The algo-
the maximum and minimum grid sizes are generated. For
rithm performance is tested in five HIV-1 protease-ligand
flexible docking, we also generated the initial population
complexes with known three-dimensional structures. In all
using a Cauchy distribution. The individual translational
five tested complexes the receptor structure is assumed to
genes are generated by adding a random perturbation
be rigid. All ligands tested are highly flexible, having more
(drawn from a Cauchy distribution) to the grid center coor-
than 10 conformational degrees of freedom.
dinates. In this way individuals are generated with higher
probability near the grid center, while still permitting that
individuals be generated far from the center. The Cauchy
distribution is given by:
In the implemented SSGA the individual chromo-
some has three genes representing the ligand translation,
four genes representing the ligand orientation and the other
π(β2 + (x−α2 ))
genes representing the ligand conformation. The
0 − ∞ < x< ∞
translational genes are the X, Y, Z reference atom coordi-
nates (usually the closest atom to the ligand center of mass),
where α and β are Cauchy distribution parameters. In this
the orientational genes are a quaternion (Maillot, 1990)
work we used α = 0 and β = 0.75. For genes corresponding
constituted by a unit vector and one orientational angle. The
to angles (dihedrals and/or orientationals), random values
Genetic algorithms for flexible docking
ranging from 0° to 360° are generated. Finally, for the
flexible docking, initially one randomly decides if a
genes corresponding to the orientational unit vector, ran-
conformational gene will be mutated or not. Then a gene in
dom values between -1 and 1 are used. The individuals are
the chosen group (conformational or not) is randomly se-
evaluated, and then are selected to suffer recombination or
lected for mutation. In this way, the seven translational/
mutation. A rank-based selection scheme (Whitley, 1995)
orientational genes have the same probability of being mu-
was used. A new individual is inserted in the population if
tated as the conformational ones.
its fitness is better than the fitness of the worst individual in
the population. The algorithm evolves until the maximum
number of the energy evaluations is reached. The reproduc-
tion operators used are classical two-point crossover and
We tested the algorithm with five HIV-1 protease-
non-uniform mutation operators (Michalewicz, 1992). The
ligand complexes where the structures were obtained from
non-uniform mutation operator, when applied to an indi-
the Protein Data Bank (PDB ID 1bve, 1hsg, 1ohr, 1hxw,
vidual i at generation ngen, mutates a randomly chosen
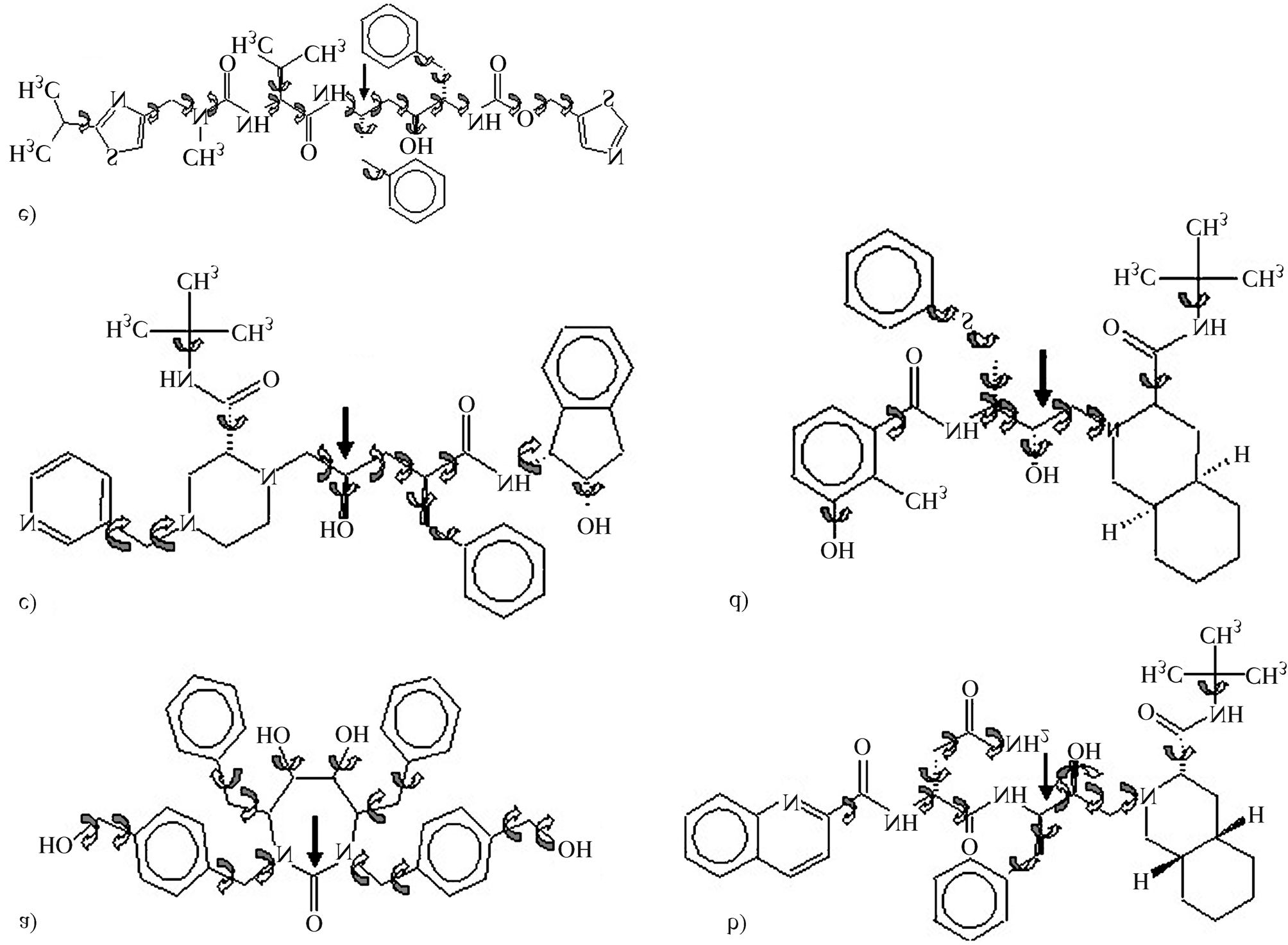
1hxb). The ligands tested are shown in Figure 1. The lig-
ands tested have conformational degrees of freedom rang-
i according to the following:
ing from 12 to 20 dihedral angles. The DMP323 ligand in
ci + ∆(ngen, bi −ci ), if τ = 0
the HIV-1 protease active site is shown in Figure 2. The
= ci +∆(ngen, ci −ai), if τ =1
grid was centered in the protein active site and we used a
grid dimension of 23 Å in each direction and a grid spacing
of 0.25 Å. The algorithm success is measured by the RMSD
i ∈ (a i , b i ), ∆(ngen, y) = y(1- r
(root mean square deviation) between the crystallographic
where ai and bi are respectively the lower and upper bounds conformation (from the corresponding PDB file) and the
for the variable ci, τ is randomly chosen as 0 or 1, r is ran- conformation found by the algorithm. A structure with a
domly chosen in [0,1] and the parameter b set to 5. In the
RMSD less than 2 Å is classified as docked and it is consid-
Figure 1 - HIV-1 protease ligands: (a) DMP323, (b) Saquinavir, (c) Indinavir, (d) Nelfinavir and (e) Ritonavir. The ligands' dihedral angles are shown by
curved arrows. The right arrows show the ligands' reference atom. The more internal dihedral angles are the neighbors' angles to the reference atom.
Magalhães et al.
and 0.7 for non-uniform mutation. The results are shown in
In flexible docking tests, all terms of the energy are
considered. We use a population of 1,000 individuals,
1,000,000 energy evaluations, and probability of 0.3 for
two-point crossover and 0.7 for non-uniform mutation. We
first tested flexible docking for DMP323 ligand with 10 and
then with 14 dihedral angles (Table 2). The results for
DMP323 flexible docking with and without the Cauchy
distribution are shown in Table 2. For all other ligands, we
used the same parameters together with the Cauchy distri-
bution. The results are shown in Table 3. We also fixed two
(three for the Ritonavir ligand) more internal dihedral an-
gles (Figure 1). The results are shown in Table 4.
Figure 2 - The DMP323 ligand in the HIV-1 protease active site.
In the rigid docking analyses, satisfactory results
ered a very good result. A structure with a RMSD less than
were found. For all ligands tested the mean RMSD ranged
3 Å is classified as partially docked. The success rate is the
from 0.046 Å to 0.099 Å. This is considered a very good re-
number of conformations found with RMSD less than 2 Å
sult in docking problems. The SSGA was able to find the
corresponding crystallographic conformation in all 10 runs
In rigid docking tests, we fixed the ligand dihedral an-
for all ligands tested, with a success rate of 100%.
gles in the position of the crystallographic structure for all
In the DMP323 flexible docking analyses, we can see
ligands, and only translational and orientational move-
that the inclusion of only four additional dihedral angles
ments are applied to the molecule. The individual chromo-
(Table 2) can interfere directly in the algorithm perfor-
some has only the translational and orientational genes, and
mance, decreasing the success rate from 100% to 30%, and
the last two terms are not evaluated for the energy function.
increasing the mean RMSD from 0.373 Å to 6.812 Å. How-
We use a population of 500 individuals, 200,000 energy
ever, with the use of the Cauchy distribution in the initial
evaluations, and probability of 0.3 for two-point crossover
population the success rate returned to 100% and with a
Table 1 - Rigid docking results.
Energy of lowest rmsd
Success ratio4 (%)
1Energy (kcal/mol) and rmsd (Å); 2The parameters used were 10 runs, 500 individuals, 200,000 energy evaluations, two-point crossover (prob. = 0.3) and
non-uniform mutation (prob. = 0.7); 3Mean in 10 runs; 4Percent of conformations found by the algorithm with rmsd < 2 Å. Standard deviations are given
in parentheses.
Table 2 - DMP323 flexible docking results.
Initial population distribution
Dihedral angles considered
Energy of lowest rmsd
Success ratio4 (%)
1Energy (kcal/mol) and rmsd (Å); 210 runs, 1,000 individuals, 1.0 x 106 energy evaluations, two-point crossover (prob. = 0.3) and non-uniform mutation
(prob. = 0.7); 3Mean in 10 runs; 4Percent of conformations found by the algorithm with rmsd < 2 Å. Standard deviations are given in parentheses.
Genetic algorithms for flexible docking
mean RMSD of 0.596 Å, with only 1,000,000 energy eval-
there is a major dependence among the dihedral angles. In
uations. This is a very good result considering that all 14 di-
this sense we observed (see Table 4) that the more internal
hedral angles are being considered, and that current
dihedral angles are critical. This seems to be due to the fact
docking programs use about 1,500,000 energy evaluations
that small variations in internal dihedral angles may cause
even in ligands with less conformational degrees of free-
larger motions in the molecule than variations in the other
dom (Morris et al., 1998). For all ligands tested the SSGA
more external dihedral angles.
was able to find the corresponding crystallographic struc-
The results obtained show the difficulty in dealing
ture with RMSD less than 2 Å at least once in 10 runs. We
with highly flexible ligands, i.e., containing many
obtained a mean RMSD ranging from 3.585 Å to 5.755 Å
conformational degrees of freedom. Moreover, the en-
and a success rate ranging from 10% to 30% in finding
closed active site of the HIV-1 protease is a considerable
docked structures, and 10% to 60% in finding partially
challenge for a docking program (Gehlhaar et al., 1995).
docked structures (Table 3). When we fixed two (three for
The EPDOCK program had a success rate of 34% in find-
the Ritonavir ligand) more internal dihedral angles (Figure
ing the corresponding crystallographic structure of the
1) we found better results (Table 4). We obtained a mean
AG-1343 HIV-1 protease ligand, with nine conformational
RMSD ranging from 1.449 Å to 3.733 Å and a success rate
degrees of freedom (Gehlhaar et al., 1995). Current dock-
ranging from 20% to 90% in docked structures, and 50% to
ing programs present a decreasing performance with the in-
90% in partially docked structures, with 10 to 17 ligand di-
creasing number of conformational degrees of freedom
hedral angles. The superior performance of DMP323, when
considered (McConkey et al., 2002). The implemented
compared to the others ligands, may be due to a minor de-
SSGA demonstrated a good performance in docking rigid
pendence among its dihedral angles and to the fact that its
ligand molecules to molecular targets in a few minutes (us-
correct conformation is placed in the center of the protein
ing a Pentium III 800 MHZ), and may be used for screening
active site; that is, privileged by using a Cauchy distribution
compounds in large databases. The flexible docking meth-
to generate the initial population. The other ligands have a
odology needs to be improved. This may be done by de-
more "open" geometry with larger arms and consequently
signing new problem-specific operators that take into
Table 3 - Flexible docking results using the Cauchy distribution.
Energy of lowest rmsd
Success ratio4 (%)
Success ratio (partially
docked structures)5 (%)
1Energy (kcal/mol) and rmsd (Å); 210 runs, 1,000 individuals, 1.0 x 106 energy evaluations, two-point crossover (prob. = 0.3) and non-uniform mutation
(prob. = 0.7); 3Mean in 10 runs; 4Percent of conformations found by the algorithm with rmsd < 2 Å; 5Percent of conformations found by the algorithm with
rmsd < 3 Å. Standard deviations are given in parentheses.
Table 4 - Flexible docking results using the Cauchy distribution without the more internal dihedral angles.
Dihedral angles considered
Energy of lowest rmsd
Success ratio4 (%)
Success ratio (partially
docked structures)5 (%)
1Energy (kcal/mol) and rmsd (Å); 210 runs, 1,000 individuals, 1.0 x 106 energy evaluations, two-point crossover (prob. = 0.3) and non-uniform mutation
(prob. = 0.7); 3Mean in 10 runs; 4Percent of conformations found by the algorithm with rmsd < 2 Å; 5Percent of conformations found by the algorithm with
rmsd < 3 Å. Standard deviations are given in parentheses.
Magalhães et al.
account critical factors of the problem such as the motion of
Holland JH (1975) Adaptation in Natural and Artificial Systems.
more internal dihedral angles. The use of a Cauchy distribu-
University of Michigan Press, Ann Arbor, MI.
tion in the initial population improved the algorithm perfor-
Jones G, Willett P, Glen RC, Leach AR and Taylor R (1997) De-
mance in all cases, but only obtained a very good result
velopment and validation of a genetic algorithm for flexible
with the DMP323 ligand. With the other ligands the im-
docking. J Mol Biol 267:727-748.
provement was not very significant, requiring the develop-
Magalhães CS, Barbosa HJC and Dardenne LE (2004) Selec-
ment of better docking strategies (Magalhães et al., 2004).
tion-insertion schemes in genetic algorithms for the flexible
ligand docking problem. Lecture Notes in Computer Sci-
ence, Springer Verlag, Berlim, 3102:368-379.
Maillot PG (1990) In Graphics Gems. Glassner AS (ed), Aca-
This work was supported by CNPq (grant number
demic Press, London, pp 498.
40.2003/2003.9) and FAPERJ (grant number E26/171.401/
Marrone TJ, Briggs JM and McCammon (1997) Structure-based
01). The authors would like to thank the Carcará Project at
drug design. Annu Rev Pharmacol Toxicol 37:71-90.
LNCC for computational resources.
McConkey BJ, Sobolev V and Edelman M (2002) The perfor-
mance of current methods in ligand-protein docking. Cur-
rent Science 83:845-855.
Michalewicz Z (1992) Genetic Algorithms + Data Structures =
Arora N and Jayaram B (1997) Strength of hydrogen bonds in σ
Evolution Programs. Springer-Verlag, New York, pp 87-88.
helices. J Comp Chem18:1245-1252.
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew
Brooks III CL, Karplus M and Pettitt BM (1988) Proteins: A theo-
RK and Olson AJ (1998) Automated docking using a
retical perspective of dynamics, structure, and thermody-
Lamarckian genetic algorithm and an empirical binding free
namics. Advances in Chemical Physics Vol LXXI. John
energy function. J Comp Chem 19:1639-1662.
Wiley & Sons, New York.
Pascutti PG, Mundim KC, Ito AS and Bisch PM (1999) Polariza-
Carlson HA and McCammon JA (2000) Accommodating protein
tion effects on peptide conformation at water-membrane in-
flexibility in computational drug design. Molecular Pharma-
terface by molecular dynamics simulation. J Comp Chem
cology 57:213-218.
Diller DJ and Verlinde CLMJ (1999) A critical evaluation of sev-
Rarey M, Kramer B, Lengauer T and Klebe G (1996) A fast flexi-
eral global optimization algorithms for the purpose of mo-
ble docking method using an incremental construction algo-
lecular docking. J Comp Chem 20:1740-1751.
rithm. J Mol Biol 261:470-489.
Ewing TJA and Kuntz ID (1997) Critical evaluation of search al-
Smith LJ, Mark AE, Dobson CM and van Gunsteren WF (1995)
gorithms for automated molecular docking and database
Comparison of MD simulations and NMR experiments for
screening. J Comp Chem 18:1175-1189.
hen lysozyme. Analysis of local fluctuations, cooperative
Gane PJ and Dean PM (2000) Recent advances in structure-based
motions, and global changes. Biochemistry 34:10918-
rational drug design. Current Opinion in Structural Biology
Wang J, Kollman PA and Kuntz ID (1999) Flexible ligand dock-
Gehlhaar DK, Verkhivker G, Rejto PA, Fogel DB, Fogel LJ and
ing: A multistep strategy approach. Proteins 36:1-19.
Freer ST (1995) Docking conformationally flexible small
Whitley D, Beveridge R, Graves C and Mathias K (1995) Test
molecules into a protein binding site through evolutionary
driving three genetic algorithms: New test functions and
programming. In: Proceedings of the Fourth Annual Confer-
geometric matching. Journal of Heuristics 1:77-104.
ence on Evolutionary Programming, MIT Press, Cambridge.
van Gunsteren WF and Berendsen HJC (1987) Groningen Molec-
Goldberg DE (1989) Genetic Algorithms in Search, Optimization
ular Simulation (GROMOS) Library Manual. Biomos, Gro-
and Machine Learning. Addison-Wesley, New York.
Source: http://www.lncc.br/~hcbm/GMB27-4-605-610-2004.pdf
A Novel, Game Theory-Based Approach to Better Understand Incentives and Stability in Clusters Endre Gedai, László Á. Kóczy, Zita Zombori László Á. Kóczy Gerd Meier zu Köcker, Institute for Innovation and Technology, iit (Berlin) Thomas A. Christensen Danish Ministry for Science, Technology and Innovation, DASTI (Copenhagen) Copenhagen, Berlin 2012 Countries all over the world look for ways to increase their competitiveness. The contribution of cooperating com-
F. L. Hellweger et al., Annals of Environmental Science / 2011, Vol 5, 61-66 APPLICABILITY OF STANDARD 1. INTRODUCTION ANTIBIOTIC TOXICITY TESTS Antibiotics, used extensively for human medicine and TO THE AMBIENT AQUATIC agriculture, enter the aquatic environment via ENVIRONMENT wastewater and other sources, where they have been found at measurable concentrations [1,2]. There may