98-071
Published May 3, 1999
Inhibition of T Cell Proliferation by Macrophage
Tryptophan Catabolism
By David H. Munn,*‡ Ebrahim Shafizadeh,* John T. Attwood,*Igor Bondarev,* Achal Pashine,* and Andrew L. Mellor*
From the *
Institute of Molecular Medicine and Genetics and the ‡
Department of Pediatrics, Medical College of Georgia, Augusta, Georgia 30912
We have recently shown that expression of the enzyme indoleamine 2,3-dioxygenase (IDO) dur-ing murine pregnancy is required to prevent rejection of the allogeneic fetus by maternal T cells.
In addition to their role in pregnancy, IDO-expressing cells are widely distributed in primaryand secondary lymphoid organs. Here we show that monocytes that have differentiated underthe influence of macrophage colony-stimulating factor acquire the ability to suppress T cell pro-liferation in vitro via rapid and selective degradation of tryptophan by IDO. IDO was induced
in macrophages by a synergistic combination of the T cell–derived signals IFN-g and CD40-ligand. Inhibition of IDO with the 1-methyl analogue of tryptophan prevented macrophage-mediated suppression. Purified T cells activated under tryptophan-deficient conditions wereable to synthesize protein, enter the cell cycle, and progress normally through the initial stagesof G1, including upregulation of IL-2 receptor and synthesis of IL-2. However, in the absenceof tryptophan, cell cycle progression halted at a mid-G1 arrest point. Restoration of tryptophan
to arrested cells was not sufficient to allow further cell cycle progression nor was costimulationvia CD28. T cells could exit the arrested state only if a second round of T cell receptor signal-ing was provided in the presence of tryptophan. These data reveal a novel mechanism by whichantigen-presenting cells can regulate T cell activation via tryptophan catabolism. We speculate
that expression of IDO by certain antigen presenting cells in vivo allows them to suppress un-wanted T cell responses.
macrophage • indoleamine 2,3-dioxygenase • T cells • tryptophan • macrophage
Certain macrophages (Møs)1 and possibly other subsets
In the course of our studies, we found that MCSF-derived
of APCs suppress T cell responses (1, 2). Immunosup-
Møs were capable of rapidly and selectively depleting the
pressive APCs have been hypothesized to play an important
essential amino acid tryptophan from cocultures and that
role in maintaining peripheral T cell tolerance. We have
this depletion occurred only in response to attempted T cell
previously shown that Møs that differentiate in vitro
un-
activation. Møs are known to possess the inducible trypto-
der the influence of macrophage colony-stimulating factor
phan-degrading enzyme indoleamine 2,3-dioxygenase (IDO),
(MCSF) acquire the ability to suppress T cell proliferation
which catalyzes the initial and rate-limiting step in the metab-
(3, 4). This attribute was not constitutively present but
olism of tryptophan along the kynurenine pathway (5–8).
It
rather was invoked only in response to attempted T cell ac-
has been postulated that the role of IDO is to inhibit prolif-
tivation. Suppressor activity was restricted to specific Mø
eration of eukaryotic intracellular pathogens (9–13) or tu-
phenotypes (e.g., the phenotype produced by MCSF), with
mor cells (14) by depriving them of tryptophan. At the
other phenotypes supporting normal T cell activation (3).
time of this study, however, no role had been proposed for
Taken together, these characteristics suggested that the in-
IDO in regulating T cell responses. Recently, we have re-
hibitory properties of MCSF-derived Møs might reflect a
ported that IDO expression in placenta is critically involved
physiologic system for regulating T cell activation. How-
in preventing rejection of the allogeneic fetus by maternal
ever, the mechanism of this inhibition was unknown.
T cells (15). The current study tests the hypothesis thattryptophan depletion via IDO is the mechanism by which
MCSF-derived Møs inhibit T cell activation in vitro and
Abbreviations used in this paper: CD40L, CD40 ligand; IDO, indoleamine
identifies a tryptophan-sensitive cell cycle arrest point dur-
2,3-dioxygenase; Mø, macrophage; MCSF; macrophage colony-stimulat-ing factor.
ing T cell activation.
J. Exp. Med. The Rockefeller University Press • 0022-1007/99/05/1363/10 $2.00Volume 189, Number 9, May 3, 1999 1363–1372http://www.jem.org
Published May 3, 1999
Materials and Methods
Recombinant human MCSF was the gift of Genet-
Validation studies showed this assay to be linear in the range of
ics Institute, Cambridge, MA. Recombinant human IFN-g was
0.1–100 mM, with an estimated threshold sensitivity of 0.05 mM.
the gift of Genentech, South San Francisco, CA. Recombinant
Where it was desirable to show that tryptophan depletion in
human CD40 ligand (CD40L) homotrimer was the gift of W.
cultures was due to IDO activity, culture supernatants were as-
Fanslow, Immunex Corp., Seattle, WA. The IDO inhibitor
sayed by HPLC for the presence of kynurenine. IDO catalyzes
1-methyl-d,l-tryptophan (16) was purchased from Aldrich Chem-
the oxidation of tryptophan to
N-formylkynurenine, which in
ical Co. 6-nitro-tryptophan (17) was synthesized by D. Boykin,
Møs is rapidly converted into kynurenine (22) and then to other
Georgia State University, Atlanta, GA, using a modification of
downstream metabolites (7). With the exception of tryptophan
the method of Moriya et al. (18). Polyclonal antiserum against
oxygenase, which is found only in hepatocytes, IDO is the only
human IFN-g was obtained from Biosource International. All
enzyme capable of degrading tryptophan along the kynurenine
other reagents were obtained from Sigma Chemical Co. unless
pathway (8). Thus, the appearance of kynurenine in cultures was
unambiguous evidence of functional IDO activity. However, be-
Cell Isolation and Culture.
Human peripheral blood mono-
cause kynurenine can be converted into other downstream me-
cytes and lymphocytes were isolated from healthy volunteer do-
tabolites, this assay was not quantitative. Where quantitative data
nors by leukocytapheresis and counterflow centrifugal elutriation,
were required, the tryptophan depletion assay described above
following appropriate informed consent under a protocol ap-
proved by our Institutional Review Board. Monocytes (.95%
HPLC assays were performed by the Medical College of
purity by cell surface markers) were cultured in 96-well plates as
Georgia Molecular Biology Core Facility. Samples were prepared
previously described (4) using RPMI 1640 with 10% newborn
by extracting 150 ml culture supernatant with 1 ml methanol.
calf serum (Hyclone) plus MCSF (200 U/ml).
Precipitated proteins were removed by centrifugation and the su-
T cell activation studies in cocultures were performed as previ-
pernatant dried under vacuum. Samples were resuspended in 100 ml
ously described (4), using the above medium supplemented with
initial mobile phase (deionized water) and an aliquot injected
an additional 5% FCS. In brief, Møs (5 3 104 cells/well) were al-
onto a C-18 column (Phenomenex Luna C-18; 250 3 4.6 mm;
lowed to differentiate for 4–6 d in MCSF, and then autologous
5 mm). Samples were eluted with a linear gradient of acetonitrile
lymphocytes (2 3 105 cells/well) were added along with mito-
in water (0–80% over 20 min), and absorbance was measured at
gen. The mitogens used in this study were anti-CD3 mAb (100
254 nm. Standards for tryptophan, kynurenine, and 1-methyl-
ng/ml, clone OKT3; American Type Culture Collection) and
tryptophan were run with each assay to establish retention times.
staphylococcal enterotoxin B (5 mg/ml; Sigma Chemical Co.).
In preliminary validation studies, the identity and purity of each
Both gave equivalent results; the data shown are from anti-CD3
peak was confirmed by mass spectroscopy.
unless otherwise specified. T cell proliferation was assessed by
Protein Synthesis and Amino Acid Analysis.
Total protein syn-
standard thymidine incorporation assay as described (3). When
thesis was measured as incorporation of tritiated leucine (4 mCi/ml)
T cell activation was studied without Møs, fresh autologous
over 24 h. TCA-insoluble proteins were precipitated and washed
monocytes were added (1:4) as nonsuppressive accessory cells.
three times in 5% TCA, and the precipitate was analyzed by liquid
Conditioned medium from cocultures of T cells and Møs was
scintillation counting. Amino acid concentrations in culture su-
prepared by harvesting supernatant 48 h after T cell addition.
pernatants were measured by HPLC in our clinical Neonatal Nu-
Conditioned medium was then used to support a second round
trition Laboratory.
of T cell activation. Mitogen and other additives were prepared
Møs were harvested with EDTA and total RNA
in tryptophan-free buffers.
prepared. Sample RNA (1 mg) was reverse transcribed with avian
A chemically defined, serum-free medium (19) selectively de-
myeloblastosis virus (AMV)-RT, and a 182-bp fragment amplified
ficient in tryptophan was prepared using tryptophan-free RPMI
with the following primers: forward, bp 237–254 of the pub-
1640 (Select-amine kit; GIBCO BRL) supplemented with in-
lished sequence (23); reverse, bp 402–418, spanning exons 3–4.
sulin (10 mg/ml), iron-saturated transferrin (5 mg/ml), and BSA
Product formation was assessed by agarose gel electrophoresis and
(1 mg/ml ultra-pure grade; measured concentration of free trypto-
ethidium bromide staining. PCR product was isolated from the
phan ,5 nM). Preliminary validation experiments confirmed that
gel and reamplified with internal primers to confirm specificity.
T cell proliferation in this medium was undetectable but was
Flow Cytometry.
Two-color FACS® analysis was performed
comparable to serum-based medium when tryptophan was
using directly conjugated mAbs as previously described (24). T
added. To study T cells in the absence of Møs, T cells were acti-
lymphocytes were identified by gating on CD3-positive cells, and
vated using anti-CD3 mAb adsorbed onto plastic tissue culture
expression of CD69, CD25, and CD71 was measured in the sec-
wells (0.5 mg/cm2 in bicarbonate buffer, pH 9) plus soluble anti-
CD28 mAb (1 mg/ml; PharMingen).
Experiments for all figures were replicated at least
Tryptophan and IDO Assays.
The tryptophan-degrading activ-
three times, and representative data are shown. Data points were
ity of Møs reflects a multifactored combination of IDO expres-
measured in triplicate and the mean reported. Error bars show
sion, tryptophan transport into the cells, and intracellular condi-
standard deviation. Where SD was ,10%, error bars have been
tions that posttranslationally affect enzyme activity (20). Therefore,
omitted for clarity. Comparisons of multiple groups within a sin-
when tryptophan depletion was the outcome of interest, we mea-
gle experiment were by ANOVA.
sured the rate of disappearance of tryptophan from culture super-natants over time. Tryptophan was assayed using the method ofBloxam and Warren (21). Proteins were precipitated with 10%
TCA and free tryptophan assayed after conversion to norharmanusing formaldehyde and FeCl
Cocultures of Møs and T Cells Are Selectively Depleted of
3. The reaction product was mea-
sured spectrofluorometrically (excitation 360 nm, emission 460
Supernatants were harvested from cocultures
nm) and compared against a standard preparation of tryptophan.
of Møs and mitogen-activated T cells after 48
h. Fresh
Regulation of T Cells by IDO
Published May 3, 1999
lymphocytes were suspended in conditioned medium andactivated with additional mitogen. Fig. 1 shows that condi-tioned medium completely failed to support T cell prolifer-ation (,1% of the proliferation in fresh medium). How-
Figure 2.
Dose–response rela-
ever, the addition of tryptophan to conditioned medium
tionship to tryptophan for T cell
fully restored its ability to support T cell proliferation, indi-
proliferation. Tryptophan was ti-
cating that tryptophan was the only component that had
trated in coculture-conditioned
been depleted. Consistent with this finding, amino acid
medium (prepared as described inFig. 1) and proliferation of T cells
analysis of conditioned media showed that all other es-
measured after 72 h.
sential amino acids were present, and only tryptophan wasundetectable (data not shown). Titration of reagent trypto-
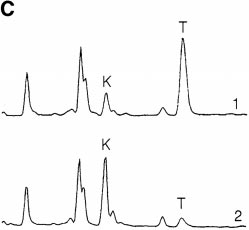
To confirm the presence of IDO activity, culture superna-
phan into conditioned medium gave a half-maximal con-
tants were assayed for kynurenine. As shown in Fig. 3 C,
centration for T cell proliferation of 0.5–1 mM (Fig. 2),
depletion of tryptophan was accompanied by a correspond-
compared with a measured concentration of tryptophan in
ing increase in kynurenine production, confirming the pres-
coculture-conditioned medium of ,50 nM (the detection
ence of functional IDO activity.
limit of our assay). Control-conditioned media from Møs
Inhibition of IDO Prevents Mø-mediated Suppression of T
alone, from cocultures of Møs 1 T cells without mitogen,
We next asked whether pharmacologic inhibition
or from T cells activated with fresh monocytes instead of
of IDO could prevent suppression of T cells in cocultures.
Møs all supported T cell proliferation comparably to freshmedium (90–140% of control; n 5 3–4/group).
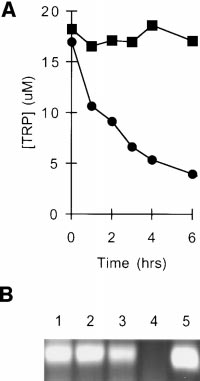
Expression of IDO by MCSF-derived Møs.
tryptophan elimination were measured by coincubatingMøs and T cells with mitogen for 24 h to allow upregula-tion of the tryptophan depletion pathway and then addingfresh tryptophan and following its disappearance. As shownin Fig. 3 A, tryptophan was eliminated by first-order kinet-
ics with a half-life of 2–3 h. The initial rate of eliminationwhen tryptophan was not limiting was up to 20,000 pmol/106 cells/h. This far exceeded the consumption attributableto cellular metabolism (see control, Fig. 3 A), as Møs with-
Figure 3.
Elimination kinetics of
out activated T cells depleted tryptophan at a rate of 300 6
tryptophan in cocultures and expression
130 pmol/106 cells/h (cumulative measurement obtained
of IDO by MCSF-derived Møs. (A)
over 7 d; data not shown). This implied that the majority
MCSF-derived Møs were cultured for
of tryptophan depletion by activated Møs was due to an in-
24 h with autologous T cells, either with
ducible system, which we suspected was IDO.
(d) or without (j) anti-CD3 mAb. Themedium was then replaced with fresh
Consistent with this finding, abundant IDO mRNA was
medium and supernatant from replicate
detectable by RT-PCR in Møs after activation, whereas
cultures harvested at the times shown. Tryptophan concentration was as-
before activation, IDO message was undetectable (Fig. 3 B).
sayed spectrofluorometrically as described in Materials and Methods. (B)IFN-g–inducible IDO mRNA in MCSF-derived Møs. RT-PCR show-ing IDO expression in MCSF-derived Møs before (lane 4) and after (lanes1–3) activation for 24 h with recombinant IFN-g. Starting RNA for thereverse transcriptase reaction in lanes 1–3 was from 20,000, 2000, and 200activated Møs, respectively, and from 20,000 unactivated Møs in lane 4.
Figure 1.
Lane 5 shows amplification of human IDO plasmid template giving the
tioned medium is selectively de-
expected 182-bp product. (C) HPLC analysis of Mø culture supernatants
pleted of tryptophan. Human
showing degradation of tryptophan and production of kynurenine.
monocytes were allowed to dif-
MCSF-derived Møs were preactivated for 24 h with IFN-g to induce
ferentiate for 5 d in MCSF. Then,
IDO expression, and then the spent medium was replaced 90:10 with
T cells were added and activated
fresh medium. Trace 1 shows the analysis of supernatant immediately af-
with anti-CD3 mAb. Condi-
ter adding fresh medium (time 0); trace 2 shows the conditioned medium
tioned medium was harvested
24 h later. The number of Møs in these experiments was kept low so that
from cocultures after 48 h and
some tryptophan would be detectable at the end of the assay. The traces
then used to support a second
shown represent the portion of the elution gradient between 28 and 42%
round of activation with fresh T
acetonitrile (minutes 7.00–10.50), during which kynurenine (K) and
cells. Replicate cultures were sup-
tryptophan (T) appeared. The peak labels are positioned at the points at
plemented with individual amino
which the purified standards eluted, which were within 63 s of the cor-
acids to the concentrations nor-
responding sample peak. Compounds present in culture medium that also
mally found in RPMI 1640. Con-
absorbed at OD254 (unlabeled peaks) were readily resolved from tryp-
trol cultures received either fresh
tophan and kynurenine, and the T and K peaks were confirmed by mass
medium (CTL) or no supple-
spectroscopy (see Materials and Methods). The experiment shown used
ment (PBS). Proliferation was
purified Møs activated with recombinant ligands; identical results were
measured by thymidine incorpo-
obtained when Møs were activated in coculture with T cells plus mito-
ration after 72 h.
gen. One of four experiments is shown.
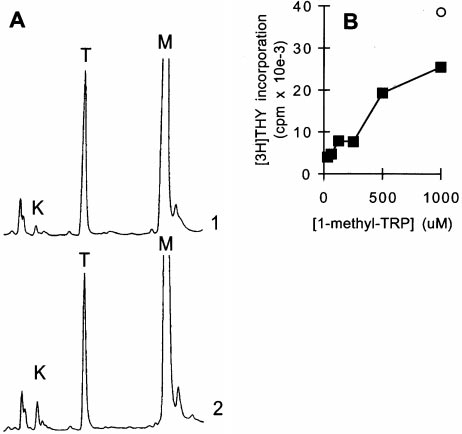
Published May 3, 1999
Figure 4.
Inhibition of IDO activity prevents Mø-mediated suppression. (A) 1-methyl-tryptophan inhibits Mø
IDO enzyme activity. MCSF-derived Møs were activated with IFN-g for 24 h to induce IDO expression, andthen fresh medium was added as described in Fig. 3, along with 1-methyl-tryptophan (1 mM). Supernatants wereanalyzed by HPLC for tryptophan (T) and kynurenine (K) immediately after the addition of fresh medium (trace1) and 24 h later (trace 2). The 1-methyl-tryptophan peak (M) is off scale at the settings used. Control cultures forthese experiments (Møs with IFN-g but without 1-methyl-tryptophan) uniformly had .90% reduction in tryp-tophan at hour 24, with a corresponding increase in kynurenine, as shown in Fig. 3. The experiment shown usedpurified Møs activated with recombinant ligands; identical results were obtained when Møs were activated in co-culture with T cells plus mitogen. (B) 1-methyl-tryptophan prevents T cell suppression in cocultures. T cells wereadded to MCSF-derived Møs and activated with anti-CD3 mAb. Replicate cultures were treated with varying
concentrations of 1-methyl-tryptophan. Proliferation was measured after 72 h by thymidine incorporation. Controls (s) show proliferation by T cellswithout Møs at the highest concentration of inhibitor used (there was no effect of inhibitor on T cells alone throughout the range of concentrations
shown). (C) A second inhibitor of IDO activity, 6-nitro-tryptophan, showed similar reversal of Mø-mediated inhibition of T cells. Experimental designas in B. (D) Supplementation with high concentrations of tryptophan prevents Mø-mediated suppression. Møs were seeded at low density (CC-lo; 5 3104 cells/well) and high density (CC-hi; 2 3 105 cells/well) and the medium supplemented with 53 the normal tryptophan concentration. Proliferationwas measured at hour 72. Controls show proliferation by Møs alone (M) and T cells alone (T).
The compound 1-methyl-tryptophan has been reported to
Tryptophan-degrading Activity Is Synergistically Induced by
be a potent competitive inhibitor of IDO activity when
Early Signals of T Cell Activation.
Møs did not degrade tryp-
tested in vitro using purified enzyme (16, 17). To deter-
tophan simply as a result of contact with T cells. Rather,
mine whether this agent could inhibit IDO activity in in-
there was an obligate requirement that the T cells attempt to
tact Møs, we added 1-methyl-tryptophan to activated Mø
activate (Fig. 3 A). In light of the existing studies implicating
cultures. As shown in Fig. 4 A, the presence of 1-methyl-
IFN-g as an inducer of IDO (25–27), we suspected that
tryptophan markedly reduced the degradation of trypto-
IFN-g from activating T cells might be the signal for IDO
phan by Møs, and this was accompanied by a correspond-
induction. Consistent with this idea, low but detectable levels
ing inhibition of kynurenine production (e.g., compare the
of IFN-g were present in cocultures within 4–6 h of T cell
ratio of tryptophan to kynurenine after 24 h in Fig. 3 C),
activation, coincident with the time that tryptophan degrada-
confirming that the target of the inhibitor was IDO.
tion began (Fig. 5 A). Neutralizing antibodies against IFN-g
Functionally, the addition of 1-methyl-tryptophan to co-
reduced the induction of tryptophan-degrading activity (Fig.
cultures abrogated the ability of Møs to suppress T cell prolif-
5 B) and reduced suppression of T cells by Møs (Fig. 5 C),
eration in a dose-dependent manner (Fig. 4 B). Although
supporting a role for IFN-g in the signaling pathway. How-
this finding was consistent with the proposed role for IDO in
ever, the dose–response relationship using recombinant
Mø-mediated suppression, it might in theory indicate an un-
IFN-g revealed that relatively high concentrations of IFN-g
anticipated immunostimulatory role for 1-methyl-tryptophan
were required for full induction of tryptophan-degrading ac-
itself. To exclude this possibility, we synthesized a second ana-
tivity (Fig. 5 D). We therefore asked whether there was an
logue of tryptophan, 6-nitro-tryptophan, which has also been
additional signal that might act in concert with IFN-g.
reported to inhibit purified IDO enzyme in vitro (17). As
CD40L is upregulated early in T cell activation and is known
shown in Fig. 4 C, 6-nitro-tryptophan also prevented Mø-
to act synergistically with IFN-g to activate other Mø func-
mediated suppression in a dose-dependent fashion (Fig. 4 C).
tions (28). Fig. 5 D shows that CD40L exerted marked syn-
Finally, we tested the effects of supplemental tryptophan on
ergy with IFN-g, shifting the dose–response curve for IFN-g
suppression. As shown in Fig. 4 D, high levels of tryptophan
one to two orders of magnitude so that significant tryptophan
did prevent suppression of T cells, provided that the number
depletion began at IFN-g concentrations of ,1 U/ml.
of Møs in cocultures was kept low. At our usual concentra-
Effect of Tryptophan Deprivation on T Cell Protein and DNA
tions of Mø, it proved impossible to supplement with suffi-
We have previously shown that T cells acti-
cient tryptophan to overcome its rapid degradation. Thus, by
vated in coculture with MCSF-derived Møs initially enter
the use of two pharmacologic inhibitors of IDO and by tryp-
the cell cycle but arrest before the first G1/S transition (4).
tophan supplementation, the mechanism of T cell suppression
We therefore asked whether a comparable phenomenon
in our system appeared to be depletion of tryptophan by IDO.
occurred when T cells were activated in the absence of
Regulation of T Cells by IDO
Published May 3, 1999
Figure 5.
IFN-g and CD40L act synergistically to induce IDO. (A) MCSF-derived Møs were cocultured with T cells and anti-CD3 mAb. Following
lymphocyte addition, culture supernatants were harvested at the times shown and assayed for IFN-g (j, left axis) and tryptophan concentration (d, rightaxis). (B) Møs and T cells were cocultured with mitogen in the presence of various concentrations of neutralizing anti–IFN-g antiserum. Tryptophan con-centration in culture supernatants was determined after 18 h. A low density of Møs was used for these experiments so as not to obscure the effect ofIFN-g. (C) Møs were cultured at a range of seeding densities as shown, and then T cells and anti-CD3 mAb were added either with (j) or without (d)neutralizing antibodies to IFN-g (100 neutralizing U/ml). Antibodies to IFN-g reduced the effectiveness of Møs in suppressing T cells, particularly whenthe number of Møs was limiting. (D) MCSF-derived Møs were cultured for 24 h with various concentrations of recombinant IFN-g, either in the pres-ence (j) or absence (m) of recombinant CD40L (500 ng/ml). At the end of the activation period, culture supernatants were assayed for the concentra-tion of tryptophan remaining. The single round point shows tryptophan degradation in response to CD40L alone.
tryptophan. Purified T cells (without monocytes or Møs)
and the time to entry into S phase determined. Control
were cultured in tryptophan-free medium using immobi-
cells, cultured with tryptophan throughout, reproducibly en-
lized anti-CD3 plus anti-CD28 mAb as activating stimuli.
tered S phase 28–32 h after initial TCR engagement (times
In this system, T cells stimulated in the presence of trypto-
are reported as 4-h ranges to reflect the limit of precision of
phan activated normally, whereas T cells stimulated with-
the assay). In contrast, T cells that had been preactivated
out tryptophan arrested before entry into the first S phase,
under tryptophan-free conditions required only 12–16 h to
as shown by the complete absence of DNA synthesis (Fig. 6).
enter S phase after tryptophan was added (Fig. 9 A), indi-
This arrest was not due to an absence of protein synthesis,
cating that significant progression through G1 had occurred
as T cells without tryptophan successfully upregulated CD69,
in the absence of tryptophan. The tryptophan-sensitive ar-
CD25 (high-affinity IL-2 receptor), and CD71 (transferrin
rest point was stable, with T cells surviving .72 h in the
receptor) (Fig. 7) and secreted IL-2 and IFN-g (Fig. 8), all
absence of tryptophan with no loss of viability. When tryp-
of which require new protein synthesis (29). Total protein
tophan was added to arrested cells, the time of entry into
synthesis, measured as incorporation of radiolabeled leucine
S phase was consistently 12–16 h, regardless of whether
during the first 24 h of activation, continued at a rate 40–
cells had been preactivated for 36, 48, or 72 h without
55% of controls (n 5 3; see Materials and Methods), despite
tryptophan. This suggested that the arrest occurred at a spe-
the absence of exogenous tryptophan. Nonetheless, no en-
cific point in G1 and that this position in the cell cycle was
try into S phase occurred. Thus, T cells activated in the ab-
maintained until tryptophan was restored.
sence of exogenous tryptophan arrested in a fashion similar
From the preceding experiments, we estimated that the
to that which we had previously observed in coculture.
tryptophan-independent portion of G1 was z14 h (calcu-
Identification of a Tryptophan-sensitive Arrest Point in
lated as the difference between the average time to S phase
The upregulation of early G1 markers suggested
for resting T cells versus the time to S phase for preacti-
that some portion of G1 was tryptophan independent. To
vated cells). To test this estimate, we deprived T cells of
test this hypothesis, T cells were activated for various times in
tryptophan during the initial 14 h of activation, then added
the absence of tryptophan, and then tryptophan was added
tryptophan just before the putative arrest point. As shownin Fig. 9 B, cultures deprived of tryptophan for the first 14 hentered S phase identically to T cells supplied with tryp-tophan throughout, supporting the hypothesis that the ini-
Figure 6.
T cells do not enter
tial portion of G1 was independent of tryptophan. In addi-
S phase in the absence of tryp-
tional experiments (not shown), delaying the addition of
tophan. T cells were activated
tryptophan beyond 14 h introduced a corresponding delay
with immobilized anti-CD3 mAb
in entry into S phase, supporting the proposed localization
plus anti-CD28, either in chemi-cally defined tryptophan-free
of the arrest point close to hour 14.
medium (j) or in the same me-
T Cells Can Commit to Cell Division in the Absence of Tryp-
dium supplemented with 25 mM
Resting (G0) T cells require TCR signaling in
tryptophan (d). DNA synthesis
order to enter G1, but subsequent progression through the
was assayed by thymidine incor-poration at the times shown.
cell cycle rapidly becomes TCR independent (for a review
Published May 3, 1999
Figure 8.
Production of IFN-g and IL-2 by T cells deprived of tryp-
tophan. T cells were activated with anti-CD3 mAb in tryptophan-freemedium (j) or in the same medium supplemented with 25 mM tryp-tophan (d) and the concentration of IFN-g (A) and IL-2 (B) in culturesupernatants determined at the times shown.
of tryptophan. As shown in Fig. 11, despite their previous48-h exposure to anti-CD3, the arrested T cells still re-quired additional TCR signaling plus the presence of tryp-tophan to exit the arrested state. Even costimulation via
CD28 was not sufficient to promote cell cycle progressionin the absence of TCR engagement.
In this study, we show that tryptophan catabolism via
IDO is the mechanism by which MCSF-derived Møs sup-press T cell proliferation in vitro. We have recently testedthis hypothesis of IDO-mediated T cell suppression in vivo
Figure 7.
Expression of activation markers on T cells deprived of tryp-
tophan. T cells were activated in tryptophan-free medium using immobi-lized anti-CD3/CD28 (heavy trace), or cultured under identical condi-tions but without anti-CD3/CD28 (light trace). At the times shown,both groups were harvested and stained for expression of activation mark-ers as described in Materials and Methods.
see reference 29). In our system, commitment to TCR-independent cell division was first detectable z6 h afterTCR engagement, and most cells were committed by hour
Figure 9.
T cells that have entered the tryptophan-sensitive arrested
12. As shown in Fig. 10, this commitment occurred identi-
state retain their position in mid-G1. (A) T cells were activated in tryp-
cally regardless of whether tryptophan was present or ab-
tophan-free medium using immobilized anti-CD3/CD28 (d). After a
sent during the relevant time period. As long as the cells
period of preactivation (24–72 h with similar results; 48 h in the experi-
were not allowed to arrest (i.e., tryptophan was supplied
ments shown), tryptophan was added and the time to entry into S phasedetermined (defined as the initiation of thymidine incorporation). Repli-
before the tryptophan-sensitive checkpoint), commitment
cate aliquots of cells were activated in tryptophan-containing medium
to cell division proceeded normally.
without the 48-h preincubation period (j). Lag time in each case was
T Cells Reverse Their Commitment to Cell Cycle Progression
defined as the time to initiation of S phase from the point at which cells
upon Entering the Arrested State.
In contrast to the experi-
saw both tryptophan and anti-CD3. The arrow shows that the lag time toS phase was shortened by 12–16 h due to preactivation in the absence of
ments shown in Fig. 10, however, once T cells entered the
tryptophan, suggesting that this portion of G1 had been accomplished be-
arrested state, simply restoring tryptophan was no longer
fore the point at which cells arrested. Representative of seven experi-
sufficient to allow cell cycle progression. T cells were acti-
ments at 36, 48, and 72 h, all showing the same lag time to S phase. (B)
vated for 48 h in tryptophan-deficient medium using im-
T cells were activated with anti-CD3/CD28 in the presence (j) or ab-
mobilized anti-CD3/CD28. The arrested cells were then
sence (d) of tryptophan. After 14 h (the time of the putative arrest pointestimated from A), tryptophan was added to the tryptophan-deficient cul-
removed from contact with anti-CD3, washed free of anti-
tures and entry into S phase determined. T cells rescued at hour 14
CD28, and transferred to medium containing normal levels
showed no delay compared with controls.
Regulation of T Cells by IDO
Published May 3, 1999
Figure 10.
phology (33–35). These IDO-expressing cells are found at
normal commitment to TCR-
several putative sites of immune tolerance or privilege, in-
independent activation in the ab-
cluding thymus, mucosa of the gut, epididymis, placenta,
sence of tryptophan. T cells wereexposed to immobilized anti-
and the anterior chamber of the eye (32, 33, 36, 37). This
CD3/CD28 for 2–12 h in the
pattern of widespread expression throughout the immune
presence (light bars) or absence
system is difficult to reconcile with a simple mechanism of
(dark bars) of tryptophan. At the
host defense. We hypothesize that IDO expression by APCs
times shown, cells were removedfrom contact with anti-CD3. Af-
functions to suppress undesirable T cell activation and thus
ter transfer, tryptophan was
helps maintain peripheral tolerance.
added to the tryptophan-defi-
Two models might be proposed by which IDO could
cient cultures, and all groups
suppress T cells in vivo: it might catalyze the production of
were continued out to hour 48.
Cells were transferred in their own conditioned medium without wash-
a suppressive metabolite of tryptophan, or it could deplete
ing and continued to receive anti-CD28 throughout. At hour 48, all
local tryptophan below some threshold level required for
groups were assayed for proliferation by thymidine incorporation. The 2-h
T cell activation. In repeated experiments, we have been
time point (no proliferation after transfer) is included as a control to con-
unable to detect any evidence of an immunosuppressive
firm that there was no carryover of anti-CD3 into the new cultures.
metabolite in coculture supernatants (Figs. 1 and 2) (4).
Furthermore, our experiments with isolated T cells imply a
using the model of allogeneic pregnancy. This model was
specific checkpoint in early T cell activation that is sensitive
chosen because it has long been recognized as paradoxical
to low concentrations of tryptophan. For these reasons, we
that the maternal immune system tolerates a genetically for-
favor the tryptophan depletion hypothesis.
eign fetus throughout gestation (30). IDO is known to be
Implicit in this hypothesis is the assumption that cells ex-
expressed in human placenta and has been reported to be
pressing IDO in vivo could create a local microenviron-
localized to the zone of contact between fetal-derived tis-
ment in which tryptophan is low, despite the availability of
sues and the maternal immune system (31). Using 1-methyl-
ample tryptophan elsewhere. In this regard, it is well estab-
tryptophan (described in Fig. 4) as a pharmacologic inhibi-
lished that delivery of a substrate into local microenviron-
tor of IDO, we have demonstrated that IDO is a required
ments is sharply limited by the rate of diffusion (Kd)
component of the mechanism by which the allogeneic fe-
through the interstitial space (38, 39). In the face of even
tus protects itself from rejection by the maternal immune
normal metabolic demands, substrate concentrations rap-
system and that inhibition of IDO breaks maternal toler-
idly fall to undetectable levels within a few cell diameters of
ance to the allogeneic fetus (15). In the same report, we also
the source of delivery (39). Because the rate of tryptophan
showed that pharmacologic inhibition of IDO enhances
consumption by IDO-expressing Møs is orders of magni-
the activation of autoreactive T cells. Thus, by two mea-
tude greater than normal metabolic demands, it is plausible
sures—breaking tolerance and enhancing autoreactivity—
that such Møs could create local conditions of very low
these data support a role for IDO in regulating T cell re-
tryptophan concentrations. Although this hypothesis is now
sponses in vivo.
speculative with regard to tryptophan, the phenomenon is
IDO has previously been viewed primarily as a host de-
well documented with regard to, for example, the local hy-
fense mechanism, inhibiting proliferation of intracellular
poxic state created within muscle tissue during exercise.
pathogens (6, 9–13) or cancer cell lines (14) by depriving
Because tryptophan degradation by IDO is much greater
them of tryptophan (for a review see reference 8). In these
than consumption by metabolic demands (Fig. 3), it is likely
settings, the proposed role of IDO has been to eliminate
that IDO constitutes the major route of tryptophan deple-
the cell's own stores of tryptophan. To our knowledge, no
tion by activated Møs. However, IDO could act in combi-
role for IDO in regulating the proliferation of adjacent cells
nation with other pathways. Møs have a high rate of pro-
has been suggested. However, both direct and indirect evi-
tein synthesis, and the incorporation of free tryptophan
dence indicates that IDO is widely expressed throughout
into proteins could contribute to local tryptophan deple-
the immune system (32, 33) and, specifically, that it is lo-
tion. Indeed, the tRNA synthetase for tryptophan (the WRS
calized to a subset of cells with a Mø or dendritic cell mor-
gene) is unique among tRNA synthetases in that it is mas-sively induced in Mø lineage cell lines (but not lymphoidlines) by the same signals that induce IDO (40). It has been
Figure 11.
T cells require TCR
signaling to exit the arrested state.
proposed that this induction allows Møs to compete prefer-
T cells were activated for 48 h in
entially for tryptophan when the concentration of substrate is
the absence of tryptophan using
low. Likewise, any pathway that transported tryptophan into
Møs, whether for protein synthesis, degradation by IDO, or
To simulate loss of contact with
incorporation into other biosynthetic pathways, would also
the APC, T cells were removedfrom the immobilized anti-CD3,
serve to deplete local tryptophan. Thus, IDO could act in
washed, and returned to culture in
concert with other catabolic pathways to render Møs an ef-
medium containing 25 mm tryp-
fective local "sink" for tryptophan.
tophan. Upon replating, replicate
The proposed tryptophan depletion model gains support
cultures received immobilizedanti-CD3, anti-CD28, or both.
from the apparent existence of a cell cycle arrest point sen-
Published May 3, 1999
sitive to tryptophan concentration. Although the absence
The requirement for a second signal from the TCR in
of any essential nutrient is, by definition, incompatible with
order to exit the arrested state is an important finding in
long-term proliferation, the arrest point we describe appears
light of our proposed biologic model. Under this model,
more specific than simple protein starvation. First, although
T cells that attempt to activate while in contact with an
protein synthesis is reduced in the absence of exogenous
IDO-expressing APC are inhibited by the local absence of
tryptophan, it still occurs at a significant rate, presumably re-
tryptophan. In theory, however, once such T cells were
flecting a combination of endogenous tryptophan stores and
committed to cell division, they could migrate elsewhere
recycling of tryptophan from catabolism of endogenous and
and complete the activation process under tryptophan-
exogenous proteins (41). Yet despite ongoing protein syn-
sufficient conditions. The data presented in Fig. 11 show
thesis, cell cycle progression is not simply delayed but rather
that once T cells have arrested, simply regaining tryptophan
is completely arrested. Second, the arrest induced by tryp-
is no longer sufficient to allow continued activation. De-
tophan deprivation occurs at a reproducible point in the cell
spite the fact that T cells would normally have become in-
cycle and remains stable once entered, suggesting a regulated
dependent of TCR signaling before the tryptophan-sensi-
process. Taken together, these attributes suggest a specific,
tive checkpoint (Fig. 10), once they enter the arrested state
tryptophan-sensitive cell cycle arrest point.
they apparently reverse this commitment and reimpose
It has been noted by several groups that deprivation of
upon themselves a requirement for a second round of TCR
certain amino acids—tryptophan in particular—exerts an
signaling. From a biologic standpoint, this would mean that
inhibitory effect on cell cycle progression that cannot be
a T cell arrested by an IDO-expressing APC would be
explained by the effect on protein synthesis (42–45). For
obliged to find a second, nonsuppressive APC presenting
that reason, it has been suggested that levels of these amino
the same antigen in order to exit the arrested state.
acids may function as specific checkpoints regulating cell cy-
What would be the fate of an arrested T cell if no such
cle progression. However, the biologic significance of such
supportive APC could be found? In vitro, we find that ar-
amino acid–specific checkpoints and the mechanism by which
rested cells undergo progressive apoptosis after several days
the levels of amino acids might be manipulated in order to
if not rescued by TCR engagement (4). Whether this means
regulate T cell activation has remained obscure. We now
that they would likewise die in vivo, enter some form of
propose a system in which regulation of local tryptophan
anergy, or return to a resting state remains to be deter-
concentration functions as a means of communication be-
mined. However, the arrested state we describe differs from
tween APCs and T cells, with APCs regulating the trypto-
classical anergy (54) in several interesting respects. First, the
phan level via IDO and T cells responding with either acti-
cells retain their responsiveness to TCR engagement (Fig.
vation or arrest, depending on the level they detect.
11). Second, costimulation via CD28 is not sufficient to
As a strategy to inhibit T cell activation, arresting pro-
rescue cells once they arrest. And third, arrested cells die if
gression through the cell cycle is not unique to tryptophan
not rescued within a relatively brief window of time. Taken
metabolism. The immunosuppressive drugs mycopheno-
together, these attributes suggest that T cells arrested by tryp-
late, rapamycin, and leflunomide all induce a mid-G1 arrest
tophan deprivation are not immediately deleted from the
in activating T cells, and this is believed to account in
repertoire but that they must find a permissive APC and
whole or part for their immunosuppressant action (46–48).
complete the activation process if they are to survive.
Recent evidence suggests that T cells require one or more
In conclusion, our hypothesis regarding the biologic
rounds of cell division to acquire a variety of effector func-
role of IDO-expressing APCs is that they are involved in
tions (49–51), so inhibiting proliferation may also inhibit
maintaining peripheral tolerance to self antigens. Our in
functional activity. In our system, it is currently unknown
vitro model has focused on MCSF-derived Møs as one
how T cells sense the level of tryptophan and trigger cell
example of immunosuppressive APCs, but dendritic cells
cycle arrest. Tryptophan-sensing systems in bacteria have
or other APCs that possess inducible IDO could likewise
been well described (52), but comparable systems in eu-
be immunosuppressive. We speculate that tryptophan ca-
karyotes have not yet been identified. However, mamma-
tabolism may constitute a previously unsuspected mecha-
lian genes such as tryptophan oxygenase are known to be
nism contributing to the regulation of peripheral T cell
regulated by changes in tryptophan levels (53), so such
sensing systems can be inferred to exist.
The authors thank J.-F. Tsai for expert technical assistance, T. Stoming for developing and performing theHPLC assays, and C. Rossignol and J. Bhatia for amino acid analysis.
This work was supported by the National Institutes of Health (grants K08 HL03395, R21 AI44759, andR01 HL60137 to D.H. Munn) and generous support from the Carlos and Marguerite Mason Trust.
Address correspondence to David H. Munn, Medical College of Georgia, IMMAG, Room CA-2010,Augusta, GA 30912. Phone: 706-721-7141; Fax: 706-721-8732; E-mail: [email protected]
Received for publication 29 April 1998 and in revised form 2 March 1999.
Regulation of T Cells by IDO
Published May 3, 1999
1. Fazekas de St. Groth, B. 1998. The evolution of self-toler-
log of tryptophan) are competitive inhibitors for indoleamine
ance: a new cell arises to meet the challenge of self-reactivity.
2,3-dioxygenase. Arch. Biochem. Biophys. 291:326–333.
Immunol. Today. 19:448–454.
17. Southan, M.D., R.J. Truscott, J.F. Jamie, L. Pelosi, M.J.
2. Banchereau, J., and R.M. Steinman. 1998. Dendritic cells
Walker, H. Maeda, Y. Iwamoto, and S. Tone. 1996. Struc-
and the control of immunity. Nature. 392:245–252.
tural requirements of the competitive binding site of recom-
3. Munn, D.H., and E. Armstrong. 1993. Cytokine regulation
binant human indoleamine 2,3-dioxygenase. Med. Chem.
of human monocyte differentiation in vitro: the tumor-cyto-
toxic phenotype induced by macrophage colony-stimulating
18. Moriya, T., K. Hagio, and N. Yoneda. 1975. A facile syn-
factor is developmentally regulated by interferon g. Cancer
thesis of 6-chloro-d-tryptophan. Bull. Chem. Soc. Japan. 48:
4. Munn, D.H., J. Pressey, A.C. Beall, R. Hudes, and M.R. Al-
19. Munn, D.H., and N.K. Cheung. 1987. Interleukin-2 en-
derson. 1996. Selective activation-induced apoptosis of pe-
hancement of monoclonal antibody-mediated cellular cyto-
ripheral T cells imposed by macrophages: a potential mecha-
toxicity against human melanoma. Cancer Res. 47:6600–
nism of antigen-specific peripheral lymphocyte deletion. J.
20. Sono, M., T. Taniguchi, Y. Watanabe, and O. Hayaishi.
5. Shimizu, T., S. Nomiyama, F. Hirata, and O. Hayaishi.
1980. Indoleamine 2,3-dioxygenase: equilibrium studies of the
1978. Indoleamine 2,3-dioxygenase: purification and some
tryptophan binding to the ferric, ferrous, and co-bound en-
properties. J. Biol. Chem. 253:4700–4706.
zymes. J. Biol. Chem. 255:1339–1345.
6. Carlin, J.M., E.C. Borden, P.M. Sondel, and G.I. Byrne.
21. Bloxam, D.L., and W.H. Warren. 1974. Error in the deter-
1989. Interferon-induced indoleamine 2,3-dioxygenase ac-
mination of tryptophan by method of Denckla and Dewey.
tivity in human mononuclear phagocytes. J. Leukoc. Biol. 45:
A revised procedure. Anal. Biochem. 60:621–625.
22. Yamamoto, S., and O. Hayaishi. 1967. Tryptophan pyrrolase
7. Werner, E.R., B. Bitterlich, D. Fuchs, A. Hausen, G. Reib-
of rabbit intestine: d- and l-tryptophan-cleaving enzyme or
negger, G. Szabo, M.P. Dierich, and H. Wachter. 1987. Hu-
enzymes. J. Biol. Chem. 242:5260–5266.
man macrophages degrade tryptophan upon induction by in-
23. Dai, W., and S. Gupta. 1990. Molecular cloning, sequencing
terferon-g. Life Sci. 41:273–280.
and expression of human interferon-g-inducible indoleamine
8. Taylor, M.W., and G. Feng. 1991. Relationship between in-
2,3-dioxygenase cDNA. Biochem. Biophys. Res. Commun. 168:
terferon-g, indoleamine 2,3-dioxygenase, and tryptophan ca-
tabolism. FASEB J. 5:2516–2522.
24. Munn, D.H., A.G. Bree, A.C. Beall, M.D. Kaviani, H. Sabio,
9. Pfefferkorn, E.R. 1984. Interferon g blocks the growth of
R.G. Schaub, R.K. Alpaugh, L.M. Weiner, and S.J. Gold-
Toxoplasma gondii in human fibroblasts by inducing the host
man. 1996. Recombinant human macrophage colony-stimu-
cells to degrade tryptophan. Proc. Natl. Acad. Sci. USA. 81:
lating factor in nonhuman primates: Selective expansion of a
CD161 monocyte subset with phenotypic similarity to pri-
10. Gupta, S.L., J.M. Carlin, P. Pyati, W. Dai, E.R. Pfefferkorn,
mate natural killer cells. Blood. 88:1215–1224.
and M.J. Murphy. 1994. Antiparasitic and antiproliferative
25. Koide, Y., and A. Yoshida. 1994. The signal transduction
effects of indoleamine 2,3-dioxygenase enzyme expression in
mechanism responsible for gamma interferon-induced in-
human fibroblasts. Infect. Immun. 62:2277–2284.
doleamine 2,3-dioxygenase gene expression. Infect. Immun.
11. Daubener, W., C. Mackenzie, and U. Hadding. 1995. Estab-
lishment of T-helper type 1- and T-helper type 2-like hu-
26. Dai, W., and S.L. Gupta. 1990. Regulation of indoleamine
man Toxoplasma antigen-specific T-cell clones. Immunol. 86:
2,3-dioxygenase gene expression in human fibroblasts by in-
terferon-g. J. Biol. Chem. 265:19871–19877.
12. Daubener, W., C. Remscheid, S. Nockemann, K. Pilz, S.
27. Chon, S.Y., H.H. Hassanain, and S.L. Gupta. 1996. Cooper-
Seghrouchni, C. Mackenzie, and U. Hadding. 1996. Anti-
ative role of interferon regulatory factor 1 and p91 (STAT1)
parasitic effector mechanisms in human brain tumor cells:
response elements in interferon-g-inducible expression of
role of interferon-g and tumor necrosis factor-a. Eur. J. Im-
human indoleamine 2,3-dioxygenase gene. J. Biol. Chem.
13. Nagineni, C.N., K. Pardhasaradhi, M.C. Martins, B. Detrick,
28. Stout, R.D., and J. Suttles. 1996. The many roles of CD40 in
and J.J. Hooks. 1996. Mechanisms of interferon-induced in-
cell-mediated inflammatory responses. Immunol. Today. 17:487–
hibition of Toxoplasma gondii replication in human retinal
pigment epithelial cells. Infect. Immun. 64:4188–4196.
29. Crabtree, G.R. 1989. Contingent genetic regulatory events
14. Aune, T.M., and S.L. Pogue. 1989. Inhibition of tumor cell
in T lymphocyte activation. Science. 243:355–361.
growth by interferon-g is mediated by two distinct mecha-
30. Medawar, P.B. 1953. Some immunological and endocrino-
nisms dependent upon oxygen tension: induction of tryp-
logical problems raised by evolution of viviparity in verte-
tophan degradation and depletion of intracellular nicotin-
brates. Symp. Soc. Exp. Biol. 7:320–328.
amide adenine dinucleotide. J. Clin. Invest. 84:863–875.
31. Kamimura, S., K. Eguchi, M. Yonezawa, and K. Sekiba.
15. Munn, D.H., M. Zhou, J.T. Attwood, I. Bondarev, S.J.
1991. Localization and developmental change of indoleamine
Conway, B. Marshall, C. Brown, and A.L. Mellor. 1998.
2,3-dioxygenase activity in the human placenta. Acta. Med.
Prevention of allogeneic fetal rejection by tryptophan catabo-
lism. Science. 281:1191–1193.
32. Yoshida, R., T. Nukiwa, Y. Watanabe, M. Fujiwara, F.
16. Cady, S.G., and M. Sono. 1991. 1-methyl-d,l-tryptophan,
Hirata, and O. Hayaishi. 1980. Regulation of indoleamine
b-(3-benzofuranyl)-d,l-alanine (the oxygen analog of tryp-
2,3-dioxygenase activity in the small intestine and the epi-
tophan), and b-[3-benzo(b)thienyl]-d,l-alanine (the sulfur ana-
didymis of mice. Arch. Biochem. Biophys. 203:343–351.
Published May 3, 1999
33. Moffett, J., M. Espey, and M. Namboodiri. 1994. Antibodies
tryptophan deprivation on L1210 cells in culture. Cancer Res.
to quinolinic acid and the determination of its cellular distri-
bution within the rat immune system. Cell Tissue Res. 278:
44. Brunner, M. 1973. Regulation of DNA synthesis by amino
acid limitation. Cancer Res. 33:29–32.
34. Espey, M., Y. Tang, H. Morse, J. Moffett, and M. Namboodiri.
45. Tobey, R., and K. Ley. 1971. Isoleucine-mediated regulation
1996. Localization of quinolinic acid in the murine AIDS
of genome replication in various mammalian cell lines. Cancer
model of retrovirus-induced immunodeficiency: implications
for neurotoxicity and dendritic cell immunopathogenesis.
46. Cherwinski, H.M., R.G. Cohn, P. Cheung, D.J. Webster,
Y.-Z. Xu, J.P. Caulfield, J.M. Young, G. Nakano, and J.T.
35. Espey, M., J. Moffett, and M. Namboodiri. 1995. Temporal
Ransom. 1995. The immunosuppressant leflunomide inhibits
and spatial changes of quinolinic acid immunoreactivity in
lymphocyte proliferation by inhibiting pyrimidine biosynthe-
the immune system of lipopolysaccharide-stimulated mice. J.
sis. J. Pharmacol. Exp. Ther. 275:1043–1049.
Leukoc. Biol. 57:199–206.
47. Terada, N., K. Takase, P. Papst, A.C. Nairn, and E.W. Gel-
36. Yoshida, R., Y. Urade, K. Nakata, Y. Watanabe, and O.
fand. 1995. Rapamycin inhibits ribosomal protein synthesis
Hayashi. 1981. Specific induction of indoleamine 2,3-dioxy-
and induces G1 prolongation in mitogen-activated T lympho-
genase by bacterial lipopolysaccharide in the mouse lung.
cytes. J. Immunol. 155:3418–3426.
Arch. Biochem. Biophys. 212:629–637.
48. Laliberte, J., A. Yee, Y. Xiong, and B.S. Mitchell. 1998. Ef-
37. Malina, H.Z., and X.D. Martin. 1996. Indoleamine 2,3-dioxy-
fects of guanine nucleotide depletion on cell cycle progres-
genase: antioxidant enzyme in the human eye. Graefe's Arch.
sion in human T lymphocytes. Blood. 91:2896–2904.
Clin. Exp. Ophthalmol. 234:457–462.
49. DeSilva, D.R., K.B. Urdahl, and M.K. Jenkins. 1991. Clonal
38. Casciari, J.J., S.V. Sotirchos, and R.M. Sutherland. 1988.
anergy is induced in vitro by T cell receptor occupancy in
Glucose diffusivity in multicellular tumor spheroids. Cancer
the absence of proliferation. J. Immunol. 147:3261–3267.
50. Bird, J.J., D.R. Brown, A.C. Mullen, N.H. Moskowitz,
39. Li, C.K. 1982. The glucose distribution in 9L rat brain multi-
M.A. Mahowald, J.R. Sider, T.F. Gajewski, C.R. Wang, and
cell tumor spheroids and its effect on cell necrosis. Cancer. 50:
S.L. Reiner. 1998. Helper T cell differentiation is controlled
by the cell cycle. Immunity. 9:229–237.
40. Fleckner, J., P.M. Martensen, A.B. Tolstrup, N.O. Kjeld-
51. Oehen, S., and K. Brduscha-Riem. 1998. Differentiation of
gaard, and J. Justesen. 1995. Differential regulation of the hu-
naive CTL to effector and memory CTL: correlation of ef-
man, interferon inducible tryptophanyl-tRNA synthetase by
fector function with phenotype and cell division. J. Immunol.
various cytokines in cell lines. Cytokine. 7:70–77.
41. Smith, C.B., G.E. Deibler, N. Eng, K. Schmidt, and L.
52. Babitzke, P. 1997. Regulation of tryptophan biosynthesis:
Sokoloff. 1988. Measurement of local cerebral protein syn-
trp-ing the TRAP or how Bacillus subtilis reinvented the
thesis in vivo: influence of recycling of amino acids derived
wheel. Molec. Microbiol. 26:1–9.
from protein degradation. Proc. Natl. Acad. Sci. USA. 85:
53. Knox, W.E., and A.H. Mehler. 1951. The adaptive increase
of the tryptophan peroxidase-oxidase system of liver. Science.
42. Dauphinais, C., and W. Waithe. 1977. PHA stimulation of
human lymphocytes during amino acid deprivation: protein,
54. Schwartz, R.H. 1996. Models of T cell anergy: is there a
RNA, and DNA synthesis. J. Cell. Physiol. 91:357–368.
common molecular mechanism? J. Exp. Med. 184:1–8.
43. Woolley, P.V., R.L. Dion, and V.H. Bono. 1974. Effects of
Regulation of T Cells by IDO
Source: http://fs.huntingdon.edu/biology/tdudley/kylee3.pdf
Health Literacy FEATURE When most people think of literacy they think of reading and writing skills. However, in Ireland, the National Adult Literacy Agency (NALA) is working with a broader understanding and definition of adult literacy. Here, Communications Officer with NALA, Jennifer Lynch details NALA's role in the area of health literacy and explains the implications for Irish society.
LC Determination of Isosorbide-5-Mononitrate in Human Plasma Himanshu S. Karmalkar&, Mohan M. Metku, Milind S. Bagul, Asmita C. Nimkar, Rajen D. Shah Raptim Research Limited, A-226, TTC Industrial Area, Mahape, Navi Mumbai, Maharashtra 400701, India; E-Mail: [email protected] Received: 3 June 2008 / Revised: 23 November 2008 / Accepted: 15 December 2008 LC-MS–MS [–] and GC–MS []However, no LC–UV method has been