Rajournals.in2
RA Journal of Applied Research Volume 1 Issue 1 Pages-18-43 November -2014 ISSN (e): Appli
Importance of Physicochemical Properties In Drug Discovery.
(Review Article)
Kapadia Akshay Bhupendra
Department of Pharmaceutical Sciences and Technology,
Institute of Chemical Technology,
Nathalal Parekh Marg, Matunga (East), Mumbai.
ABSTRACT:
Drug discovery is a complex and demanding
physicochemical properties in drug discovery and
enterprise. In recent years there has been a
exploratory development.
significant
discussion
discovery
This review is based on how physicochemical
developmental processes for new chemical
properties of compounds can be optimized for
entities, wherein various parameters like PK,
drug discovery.
toxicity, solubility, stability are addressed. The
'Rule of Five', gained wide acceptance as an
approach to reduce attrition in drug discovery
PROPERTIES:
and development. However, analysis of recent
Most of the drugs used in medicine behave in
trends reveals that the physical properties of
solution as weak acids, weak bases, or sometimes
molecules that are currently being synthesized in
as both weak acids and weak bases. The term
discovery
companies
"physicochemical properties" refers to the
significantly from those of recently discovered
influence of the organic functional groups within a
compounds
molecule on its acid-base properties, water
development. The consequences of the marked
solubility, partition coefficient, crystal structure,
deviation in the physicochemical properties result
stereochemistry, and so on. All these properties
in a greater likelihood of lack of selectivity and
influence the absorption, distribution, metabolism,
attrition in drug development. Tackling the threat
excretion, and toxicity of the molecule. The lead
of compound-related toxicological attrition needs
optimization stage of drug discovery usually calls
to move to the mainstream of medicinal chemistry
for specific methods that attempt to model
decision-making. The impacts of these rules on
properties such as oral absorption, blood–brain
oral absorption are discussed, and approaches
barrier penetration, distribution, metabolism and
are suggested for the prediction, assessment and
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014

RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
its toxicity effects in the individual. Many ADME
discovery projects represent major economic
models include physicochemical properties as
losses for the companies. Furthermore years and
descriptors; calculation of these properties has to
work on these discoveries and developments are
be widely studied, because success or failure of
lost. Ultimately, the introduction of a new drug
the drug candidate solely depends on the
candidate in the market is delayed. PK assessment
should be seeded in the late discovery or the
Christopher A. Lipinski has commented:
predevelopment stage. This testing succeeds in
‘Drug-like is defined as those compounds that
have sufficiently acceptable ADME properties and
sufficiently acceptable toxicity properties to
survive through the completion of human Phase I
clinical trials [Lipinski, C. A. (2000)].'
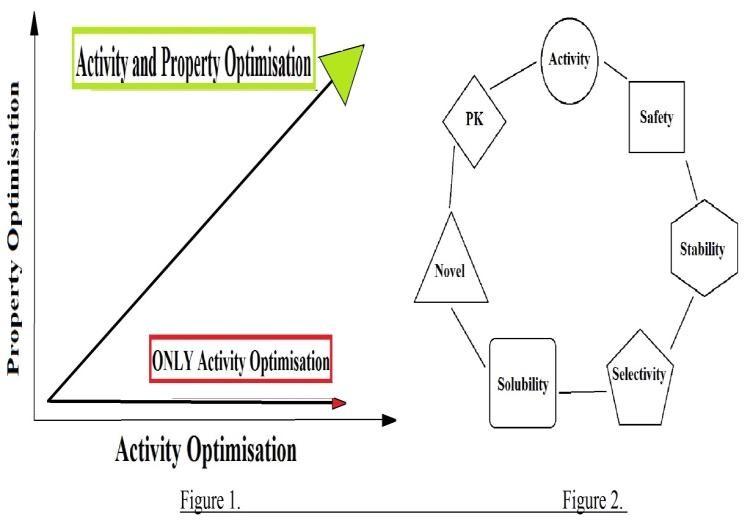
For a discovery project team it is important to
focus on both activity and properties of the
candidate [Kerns, E. H.et al. (2003)], if the focus
is solely on the activity , the team may arrive with
Figure 1. Representation showing optimisation of
a candidate whose properties are worse than the
both Activity and Property. Figure2. Juggling
HTS hit. Once a nanomolar activity is obtained it
is hard to go back and fix the structural
keeping poor candidates from progressing into
modifications because the substructure may have
development greatly reduces the rates of attrition.
to be modified again which were added in order to
Another useful anology is juggling. A proper
enhance binding affinity. Optimization of drug-
balance of crucial elements have to be maintained
like properties like absorption, distribution,
in order to achieve success.
metabolism, excretion and toxicity (ADME/T) in
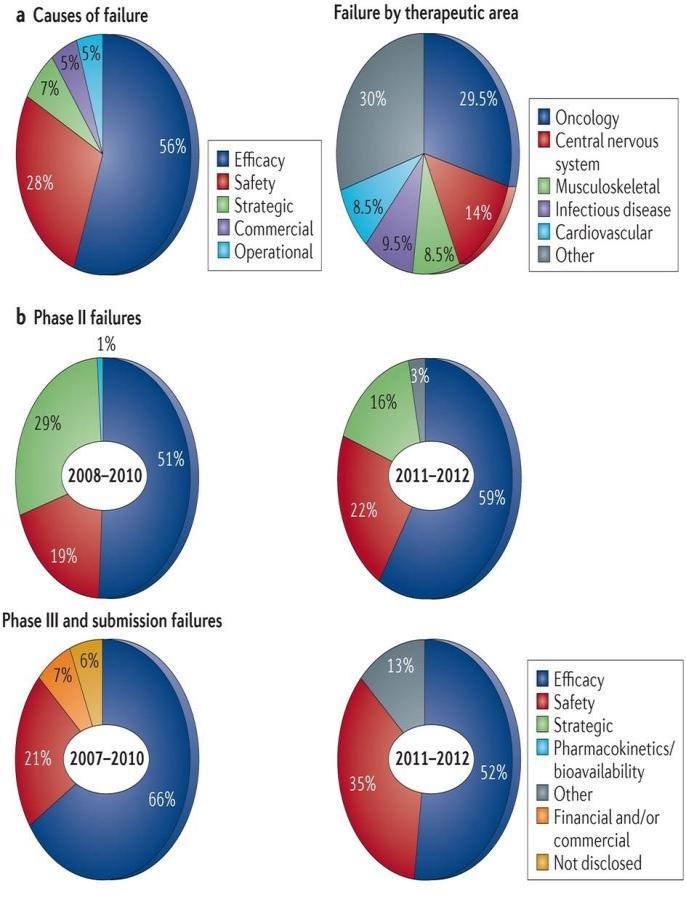
Drug attrition is an alarming situation in recent
time. A research carried out by J. Arrowsmith et
selectivity) increases drug discovery success.
al., (2013) shows that in 2011-2012, there were a
The cost of development of new chemical entities
total of 148 failures between Phase II and
is generally high wherein failures of these
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014

RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
submission (also including Phase I/II studies in
shows various parameters, causes and trends in
patients and major new indications of already
attrition rates.
marketed drugs). Of these, 105 had reported
reasons for failure. The majorities were due to a
lack of efficacy (56%) or to safety issues (28%);
here, failures that were due to an insufficient
therapeutic index were included under the safety
On comparing by phase bases, for the most recent
year range, the proportion of failures due to lack
of efficacy was higher in Phase II (59%), but still
disturbingly high in Phase III and beyond (52%).
The proportion of failures due to safety issues is
higher in Phase III and beyond compared with
Phase II at 35% and 22%, respectively, which
may be due to safety issues that only become
apparent in larger numbers of patients and/or
When the failure rates are broken down by
therapeutic area, oncology and central nervous
system (CNS) disorders account for 44% (30%
and 14%, respectively) of all the 105 failures
between Phase II and submission for which
reasons have been reported. However, almost 50%
Figure 3. Trends in attrition rates. a. Of the 148
of CNS and endocrinology (diabetes) failures (13
failures between Phase II and submission in 2011
out of 29, and 4 out of 8, respectively) are
and 2012, reasons were reported for 105; the
excluded from these numbers because the reason
majority of failures were due to lack of efficacy,
for the failure has not been disclosed. Figure3
as shown on the left. On the right, the 105
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014

RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
reported failures are broken down according to
therapeutic area. b. Comparison of the reasons for
failures in Phase II and Phase III trials in 2011 and
2012 with those in earlier periods that we reported
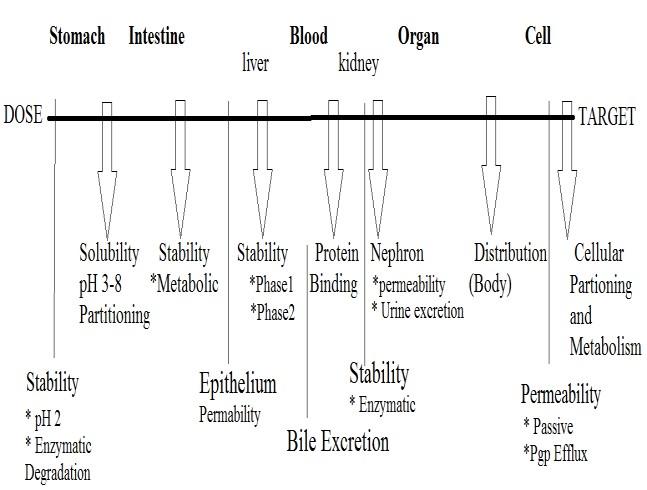
BARRIERS IN DRUG EXPOSURE:
When a drug molecule is administered it has to:
Dissolve in the biological fluids i.e. gastric fluids,
intestinal fluids, blood plasma etc.
Figure 4. Overview of Barriers in the pathway of
Survive a range of pH from 1.5 in the stomach to
Drug Delivery to the target.
8.0 until it reaches the large intestine andfurther to
Consequences of chirality on barriers and
Survive Intestinal and Gut bacteria.
properties:
Permeate through the biological membranes in the
Survive Metabolism by the enzymes.
enantiomers, diastereo-isomers exhibit different
Avoid active transport to bile.
physicochemical properties, including melting
Avoid excretion by kidneys.
Reach the target organ.
chromatographic behavior. The physicochemical
Show its therapeutic activity and
properties of a drug molecule are dependent not
selectivity towards the target receptor.
only on what functional groups are present in the
Reduce partition and binding to unwanted sites.
molecule but also on the spatial arrangement of
these groups. This becomes an especially
important factor when a molecule is subjected to
an asymmetric environment, such as the human
body. Proteins and other biological
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014

RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
macromolecules are asymmetric in nature, how a
Metabolism (Binding, orientation of molecules
particular drug molecule interacts with these
positions to the reactive moiety)
macromolecules is determined by the three-
Plasma protein binding (Binding to specific target
dimensional orientation of the organic functional
groups present. If crucial functional groups are not
Toxicity, such as CYP inhibition, hERG blocking
occupying the proper spatial region surrounding
Table 1. Effect of Sterioselectivity on Renal
interactions with the biological macromolecule (or
Clearance
receptor) will not be possible, potentially
Enantiomeric Ratio*
therapeutic effect. However, if these functional
groups are in the proper three-dimensional
orientation, the drug can produce its interaction
with the receptor. Therefore is very important for
the medicinal chemist developing a new
molecular entity for therapeutic use to understand
*ratio of renal clearance of the two enantiomers.
not only what functional groups are responsible
for the drug's activity but also what three-
dimensional orientation of these groups is needed.
Log P: It is defined as the Log of the partition co-
efficient of the compound between an organic
phase and aqueous phase at a pH where all the
pharmacodynamics properties of the molecule.
compound molecules are in the neutral form
Examples are as follows:
[Rekker et al. (1992)].
Solubility (Crystal forms of enantiomers are
The organic phase used is generally n-octanol and
the aqueous phase is unionized water. Log P
depends on the partition coefficient of the neutral
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014

RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
molecules between the two phases. Abraham et al.
affinity to the polar aqueous phase than the non-
have shown that Log P is affected by several
polar organic phase. The fraction of molecules
fundamental structural properties of the compound
ionized depends on the pH of the aqueous
[Mannhold et al (2009)]:
solution, the pKa of the compound and whether
Molecular volume: related to the molecular
the compound is an acid or a base. For bases the
weight of the compound which affects the size of
neutral/cations ratio of the molecules in solution
the cavity in the solvent to solubilize the
increases with increasing pH, hence the Log D
value increases with increasing pH. Conversely
Di-polarity: affects the polar alignment of the
for acids, the neutral/anion ratio decreases with
compound with the solvent
increasing pH, and Log D also decreases. Thus
Hydrogen bond acidity: acceptance of hydrogen
Log D is directly proportional to the neutral/ion
bonds of the solvent.
ratio of the molecules in the solution.
Hydrogen bond basicity donation of hydrogen
Parameters affecting Lipophilicity [Abraham et
bonds to the solvent.
Change in phases: Partitioning between octanol
Log D: It is defined as the Log of the distribution
and water is different than that between
co-efficient of the compound between an organic
cyclohexane and water; this is due to the
phase and aqueous phase at a specified pH (x)
molecular properties of the phases.
where the compound molecules are in the partly in
pH: Affecting the degree of ionization
the ionic form and a portion may be in the neutral
Ionic strength of the solvent: Affects polarity,
form [Hansh et al. (2004)].
molecular interactions and forms in-situ salts (as
counter ions) with drug molecules.
Co-solutes and co-solvents: May change the
partitioning behavior of molecules even in smaller
Log D depends on the partitioning co-efficient of
the neutral portion of the molecule population plus
the partioning portion of the ionized portion of the
molecular population. Ions generally have greater
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
Lipophilicity co-relations [Hansh et al. (2004)]:
Effect of Log D on optimization
Permeability: Increase in lipophilicity increases
parameters [Kerns, E. H.et al. (2010)]:
the permeability through the lipid bilayer
hence increase in Absorption:
Common impact on
Common impact
properties
Distribution: Increase lipophilicity, Increases
Permeability low due
Distribution low
Plasma protein binding.
to passive trans
Oral absorption and
cellular diffusion
If MW less than 200,
compounds occurs faster.
permeation via Para-
cellular diffusion
Elimination: Compounds are protein bound, hence
possible Metabolism low
elimination and excretion of these compounds is
Solubility moderate
Oral absorption and
Toxicity: Increased stay in the body may result
into undesirable side effects.
Oral bioavailability
Permeability high
[Lombardo et al. (2002)] also showed co-relations
Metabolism moderate
between the Volume of Distribution (Vd) and
Permeability high
lipophilicity. Increase in lipophilicity increases the
(especially amines)
plasma binding of the drug, increasing the Vd,
thus leading to increase in the retention of the
drug in the body.
Table 2. Effect of Log P on optimization
pKa indicates the ionizability of the compound. It
parameters [Kerns, E. H.et al. (2010)]:
is a function of the acidity or basicity of group(s)
Property
the logarithmic measure of the acid dissociation
constant (Ka). The logarithmic constant, pKa, is
equal to −log10 Ka.
Aqueous solubility
pKa = - log ([H+]*[A-] / [HA])
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
pH = pKa + log([B] / [HB+])
pKa = - log ([H+]*[B] / [HB+])
Thus, [HB+] / [B] = 10 (pKa – pH)
Using these relationships, concentration of the
neutral and ionic species can be calculated at any
pH, if the pKa is known. When pH equals pKa,
there is an equal concentration of ionic and neutral
species in the solution.
pKa is an important parameter because majority of
the drugs contain ionizable groups. Most of the
Figure 5. Concentration of neutral and ionic
drugs are basic, few are acidic and a minor part is
species of acids and bases at pH above and below
their pKa.
As pKa determines the degree of ionization, it has
To further simplify, acids with lower pKa value
major effect on solubility and permeability. A
are stronger because as the pH decreases there is a
particular relationship between the permeability
greater concentration of neutral acid molecules
and solubility is defined which states that they are
(HA) and a lower concentration of anionic acid
inversely proportional. For Acidic molecules,
molecules (A-) in the solution. Similarly bases
decreases with increasing pH,
with lower pKa values are weaker because as the
because as acidity decreases, ionization increases
pH decreases, there is a lower concentration of
and diffusion of anionic moiety through the
membrane becomes difficult, conversely the
concentration of cationic base molecules (HB+) in
increases as ionization increases.
solution [Kerns E H. et al. (2001)].
Similarly for bases, as the pH decreases,
5.1. The Henderson-Hasselbalch equation [Avdeef
ionization increases, permeability decreases and
et al. (2001)] is a useful relationship for
solubility increases. pKa also affects the activity
discovery.For acids:
of a structural series by showing changes in
a + log ([A-] / [HA])
interaction at the active site of the target protein
Thus, [HA/A-] = 10 (pKa – pH)
[Martin et al. (1993)].
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
SOLUBILITY:
class of compounds. E.g. Cimetidine, Nifidipine,
It is defined as the maximum dissolved solute
concentration under the given solution conditions.
Class IV: Low Solubility and Low Permeability
It determines the oral bioavailability and the
(risk-philic): development of the compounds of
intestinal absorption. Lipinski et al. stated that
this class is costly and risky. No in-vitro/in-vivo
solubility is a much larger criterion as compared
to permeability in drug discovery [Lipinski et al.
Hydrochlorothiazide, Furosemide, Tobramycin.
(2012)]. The solubility classifications used in drug
5.2. Factors that affect solubility [Rouland M.
discovery is given below [Waterbeemd H. (1998);
Wu Chi-Yuan et al. (2005)]:
Compound structure: More lipophilic, less the
polar solubility and more hydrophilic, less the
The Biopharmaceutics Classification System:
lipid solubility.
In order to promote the optimum candidate to
pKa: when the pH of the solution equals the pKa of
development and streamline the transition to
the compound its solubility is twice the intrinsic
development, the BCS was invented. It divides all
solubility of the compound.
the drug candidates into 4 classes:
Size: Larger the molecule, less its solubility.
Class I: High Solubility and High Permeability
Crystal lattice energy: Greater the energy, lesser
(amphiphilic); the most ideal class for oral
its aqueous solubility, due to stronger bonding of
the crystal lattice.
Class II: Low Solubility and High Permeability
(lipophilic); formulation manipulations are used to
Amorphous: Highly Soluble
increase the solubility of these classes of
Crystalline: Moderately Soluble
Class III: High Solubility and Low Permeability
Liquid: Polar liquids more soluble in aqueous
(hydrophilic); prodrug strategies are used for these
solutions than non-polar liquids.
Composition and physical condition of solvents:
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
Type of solvents: Polarity
solution. This has high implications for the
Amount (%) of solvents
solubility of compounds in various physiological
Solution components
fluids and solutions at different pH. Thus the
pH: Acidic compounds more soluble in Basic pH
solubility of a mono- acid or a mono-base is
temperature: Increase in temperature increases
B +H2O ↔ OH- + HB+
Medicinal chemists have the ability to alter the
At equilibrium a mono-acid and a mono-base can
solubility by manipulating the structure thus
be described as:
altering the physicochemical properties of the
empirically derived a general solubility equation
‘S' is Solubility.
for estimating the aqueous solubility of the
A mathematical derivation of the Henderson-
compound. The equation demonstrates the effect
Hasselbalch equation provides the insight for
of lipophilicity and crystal lattice energy on
solubility as under:
solubility [Yalkowsky et al. (1992)].
S = So (1 + 10(pH – pKa))
Equation: Log S = 0.8 – Log Pow – 0.01(MP – 25)
S = So (1 + 10(pKa – pH))……………where ‘So' is
Here, S is the Solubility, Log P
the Intrinsic Solubility
octanol/water partition co-efficient (measure of
lipophilicity), and MP is the melting point
exponentially with the difference pH and pKa.
(measure of the crystal lattice strength).
Examples are listed in the table. Barbital and
Thus, solubility decreases 10 fold when Log P
amobarbital have same pKa, but barbital have
increases by 1 unit or the melting point increases
much higher intrinsic solubility, because of its
extra lipophilic chain in amobarbital, thus
Therefore the solubility of a compound at a
solubility of barbital is more as compared with
particular pH is the sum of its intrinsic solubility
amobarbital. Naproxen and
i.e. the solubility of the neutral species as well as
somewhat similar intrinsic solubility, but different
the ionic species portion of the molecules in the
pKa values; hence their solubility differs
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
extensively at pH 9. As the difference in pH and
soluble in the later section of the small intestine
pKa increases solubility increases exponentially
because the region is more basic.
[Lee Y. et al. (2003)]. Examples to prove the
Table 5. Distribution of Drugs Based on the
above statement:
Physiological pH in the Body.
Table 4. Solubility at a given pH is a given
Type of drugs
function of the intrinsic solubility of the
Neutral portion of the Molecules and solubility
Small intestine
5.5-7 Basic and Neutral
of the ionized portion of Molecules [Lee Y. et
al. (2003)].
Neutral and Basic
Intrinsic
Solubility @pH9
Muscle tissue
solubility
Adipose tissue
Lipophilic drugs
Amobarbital 7.9
Barbital
5.3. Effects of solubility:
As the compound dissolves, its concentration in
the solution increases, hence its absorption occurs
at a faster rate. Compounds with low solubility
have low oral bioavailability. Cases of toxicity are
also seen with compounds showing low solubility,
due to retention of drug in the GI tract E.g.
permeability and maximum absorbable dose.
Cocaine, THC etc.
The human GI tract shows a pH gradient along its
solubility than low-permeability compounds to
length varying from strongly acidic to basic.
achieve maximum oral absorption [Bighley L.D.
Acidic and basic drugs have different solubility
throughout the GI tract. Bases are more soluble in
the stomach and the upper part of the intestine due
to ionization at acidic pH. Acidic drugs are more
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
Lipinski C A. (2000) has developed a useful
graphical co-relation of solubility, permeability
The velocity of the molecule passage through a
biological membrane barrier is known as
permeability [Goodwin J. T. et al. (2001)].
Prediction of in-vitro permeability can enhance a
wide range of drug discovery investigations, help
with understanding cell based bioassays, and
assist prediction and interpretation of in-vivo
Figure7. Graph for estimating solubility of
Discovery Compounds.
encounter several different membrane barriers in
In the above example, the compound has average
the living system [Artursson P. (2002)]. They
permeability (K
include Gastrointestinal (GI) epithelial cells,
a) and average potency ("1.0"
mg/kg considering the dose to be fully absorbed),
Blood capillary wall, Hepatocyte membrane,
the compound should have minimum stability of
Glomerulus, Restrictive organ barriers: Blood
52mcg/mL to be completely absorbed. In case of
Brain Barriers and Target cell membrane.
non-potent compounds, with a dose of about
Permeation through the membranes occurs by five
10mg/kg and having average permeability, the
major mechanisms: (a) Passive diffusion, (b)
solubility must be 10 times higher i.e.
Endocytosis, (c) Uptake transport, (d) Para-
520mcg/mL. These estimates help to provide
cellular transport and (e) Efflux transport
useful guidelines for optimization of solubility
[Brahmankar D. M. (2005); Lin J. H. (1997)].
parameter during discovery. The following is the
Lipid Bilayer Membrane
Table 7. Classification of Drugs based on
Solubility [Kerns, E. H.et al. (2010)].
Less than 10mcg/mL
Low Solubility
Moderate Solubility
More than 60 mcg/mL
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
Figure 8. Complex Model of a Lipid Bilayer
Another term that comes into play is the combined
or composite permeability, which is the result of
The phospholipid molecules assemble as a bilayer
dynamic interaction of local conditions and how
ranging approximately 490 nm in length, with the
they affect the various permeability mechanisms.
hydrophobic portion oriented inwards and the
The conditions that may result in change of these
hydrophilic phosphate heads towards the water
molecules. Molecules diffuse through this bilayer
gradient, transport affinity, molecular size and
membrane by breaking the polar hydrogen bonds
by shedding the hydrating water molecules and
BLOOD-BRAIN BARRIER (BBB):
diffuse inside, passing through the tightly packed
BBB is restrictive for many compounds due
region of the lipid chains around the glycerol
owing to the p-glycoprotein efflux, absence of
backbone and moves further to the more distorted
Para-cellular permeation and limited pinocytosis.
lipid region of the lipid aliphatic chains in the
In order the drug to be administered to the CNS or
middle of the membrane. Molecules with lower
brain tissue its permeation through the BBB
molecule weight passes through the membrane
should occur. Many of the compounds generally
more easily as compared to the higher molecular
fail in achieving the desired therapeutic efficacy
weight compounds, due to the tightly packed
due to impermeability through the BBB. There are
arrangement. Also, lipophilic molecules pass
many mechanisms or say a combination of
through the non-polar central core of the
mechanisms that limit the permeability of these
membrane more easily than the hydrophilic ones.
drugs through the BBB. The BBB is associated
Molecules then move through the other side
with the micro capillary blood vessels that run
chains and polar heads of the other side of the
throughout the brain in close proximity to the
membrane, thus regaining the polar hydrolysable
brain cells. These cells provide the necessary
water molecules and form hydrogen bonds again.
nutrients and also take away the excreted products
Membrane permeability differs from tissue to
from the brain cells. They possess a surface area
tissue, as composition of different tissues may
of about 12mm2. The BBB consist of endothelial
vary, like Gastro-Intestinal tract v/s the Blood
cells that form a monolayer lining on the inner
surface of the capillaries. The endothelial layer
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
consists if mainly astrocyte and pericyte cells,
Enhancements feature which generally increase
which do not resist drug permeation but can, alter
the uptake of nutrients like amino acids, peptides,
endothelial cell characteristics. CNS drugs must
glucose etc. and other endogenous compounds.
permeate through the endothelial cells to penetrate
Uptake enhancement is most commonly delivered
the brain cells. The mechanism through which
drugs permeate through the barrier is shown in the
Nonspecific binding to plasma proteins and lipids
in the brain tissue
8.1.Mechanisms that affect the BBB permeation
Drug molecules that permeate the BBB are subject
[Kerns E. H. et al. (2006)]:
to no specific protein binding inside the brain. The
Restricted physicochemical characters that limit
free drug hypothesis states that binding of the
passive diffusion
drug to some other substrate reduces the
Physicochemical properties considerations as
therapeutic receptor concentration and thus reduce
stated by Pardridge, as well as the compound
in activity is seen.
should have fewer hydrogen bond donors, higher
Plasma Protein binding
log P, lower PSA and a few rotatable bonds.
PPB greatly limits the permeation to the brain,
High efflux activity
because the on/off kinetic models are low to
PGp efflux limits the molecules before they can
moderate and very little drug is available permeate
reach the brain cell. Thus an efficient strategy is to
reduce efflux by PGp
Clearance of the compound from the ECF into the
Lack of sites for Para-cellular permeation and
capillary wall fenestrations
The second interface between blood and the brain
Tight junctions between the cells,
is choroid plexus. The BBB interfaces with the
Limited pinocytosis
blood and the ECF of the brain. The choroid
Endothelial cell metabolism and metabolic
plexus interfaces with the blood and the CSF an is
hence the blood cerebrospinal fluid barrier
Increase in hepatic clearance affects the amount of
drug reaching the brain
Limitations of BCSFB is
Uptake transport
Surface area is 5000 time smaller than BBB
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
There is little mixing of the components of CSF
These rules were first used by Pfizer, prior to their
publication and since then it has been widely used.
CSF flows very fast from the brain tissue toward
The rule states [Lipinski et al. (2004)]:
the arachnoid villi
Poor permeation and absorption are more likely
CSF is turned over every 5 hours.
> 5 hydrogen bond donors (expressed as the sum
Figure9. Acids Poorly Permeate BBB, whereas
of all OH and NH)
Bases have good BBB Permeability [Clark D. E.
Molecular weight > 500
>10 hydrogen bond acceptors (expressed as a sum
predominantly negatively charged phospholipids
head groups in the BBB [Liu X. (2006)]. About 75
Substrates for biological transporters are
% of the prescribed drugs are basic, 19% are
exception to the rule.
neutral and 6% are acidic.
A study conducted by Veber on rats showed,
molecular flexibility, polar surface area and
hydrogen bond count are important determinants
for oral bioavailability. Rotatable bond also
account in the picture, which may be calculated
electronically or manually. Calculation of PSA
can be done using sophisticated softwares.
PROPERTY
PROFILING
Veber rules for good bioavailability in rats [Veber
FROM STRUCTURE:
D. et al. (2002)]:
Lipinski rules:
≤ 10 rotatable bonds
The declaration of ‘The Rule of 5' as stated in the
report of Lipinski et al, opened a new way for the
≤ 12 total H bond donors and acceptors
classification of the physicochemical properties of
the drug compounds [Lipinski et al. (2012)].
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
bond donors >2. This is in agreement that
hydrogen bond donors are limiting than hydrogen
Another set of rules compiled by Clark [Clark D
E. (2003)] and Lobell [Lobell et al. (2003)]
suggests that the structure should have the
Figure 10. Application of Lipinski and Veber rule
Buspirone. Refer Table 8.
PSA < 600 – 700 nm2
Table 8. Calculations for Buspirone
Lipinski Rules
Veber Rules
Opera el al proposed set of rule of 3 for lead-like
Rotatable Bonds = 5
The ‘Rule of 3' for lead-like compounds as
Total Hydrogen Bonds = 31
proposed by Oprea [Opera et al. (2002)]:
Molecular weight ≤ 300
Good Absorption
Good Oral Bioavailability
Rotatable bonds ≤ 3
Pardridge- rules for BBB permeability:
Hydrogen bond donors ≤ 3
Physicochemical properties greatly affect BBB
Hydrogen bond acceptors ≤ 3
permeation. A set of physicochemical rules was
Polar surface area (PSA) ≤ 600nm2
first proposed by Pardridge [Pardridge (1995)].
Rules of Thumb for a Given Set Molecular
The structure of the compound should have:
Hydrogen bonds (total): < 8 -10
A set of simple, consistent structure–property
Molecular Weight < 400-500
guides have been determined from an analysis of a
No acidic moiety.
number of key ADMET assays run within GSK:
Sparklin [Maurer T S. et al. (2005)] further
solubility, permeability, bioavailability, volume of
suggested that Hydrogen bond acceptors <6 and
distribution, plasma protein binding, CNS
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
penetration, brain tissue binding, P-gp efflux,
one or more of the parameter lie above the cut-
1A2/2C9/2C19/2D6/3A4
Figure 11. Indication of How Changes in Key
models have been developed on almost all the key
Molecular Properties will affect a Range of
ADMET Parameters. a) For Neutral Molecules, b)
pharmaceuticals industry and are reviewed in
For Basic Molecules, c) for Acidic Molecules, d)
detail in many researches. Much of the research
For Amphiphilic Molecules. a Expressed relative
on in-silico ADMET and QSPR (Quantitative
to the mean value of the data sets. MWT and
Structure Property Relationship) models is based
ClogP cut-offs of 400 and 4, respectively, are
on more advanced statistical data as reported in
used. * Optimum ClogP bin is 3–5 with respect to
the literature. To counter the general reduction in
permeability. ** Average to high volumes rather
interpretability of QSPR models, an attempt was
than high, low, or average generally considered
made to demonstrate a set of simple rules of
optimum. *** Low CNS considered optimum,
thumb based on large data sets a range of ADMET
although for targets in the brain, this will be
assays run within GSK [Gleeson M. P. (2008)].
reversed. **** Some isoforms show a nonlinear
The results were compiled and a set of rules were
relationship with ClogP and/or MWT. These are
formulated wherein qualitatively predict the
ADMET issues most likely to be experienced for
a molecule based on its ClogP, MWT, and
ionization state, without the need for complex
computer simulations. The likelihood of a
molecule having a particular It is clear that almost
all ADMET parameters increase with either
increasing MWT and/or ClogP, a single combined
ClogP/MWT category has been used for
simplicity. Molecules lie in the more desirable
category if both MWT < 400 and ClogP < 4,
while they are classified as less-desirable should
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
The rather simplistic modeling used here has the
advantage of allowing scientists to make cross
comparisons between a large numbers of ADMET
assays. It then becomes easy to assess in a
qualitative fashion how changes in the key
physicochemical parameters will impact each of
the different ADMET parameters in a particular
This simplicity can be useful in a lead
optimization environment where one does not
optimize ADMET parameters in isolation. Such
simple rules could also be used in the Hit-to-Lead
stage to identify the likely ADMET issues of a
given lead, allowing resources to be more
effectively directed to the areas identified before
the molecule enters lead optimization.
These rules aid in the assessment of compounds.
They are typically used for the following
Anticipating of the drug like properties of
potential compounds i.e. lead molecules when
planning synthesis.
Evaluating the drug-like properties of compounds
being considered for purchase from a compound
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
CONCLUSION:
REFERENCES:
Over the years, strategies for reducing failure of
Book References:
lead molecules have been stated and optimization
1. Beale, John M., and John Block. Organic
of physicochemical properties has been an
medicinal and pharmaceutical chemistry.
important parameter. Figure 44 shows how
Lippincott Williams & Wilkins, 2010.
incorporation of evaluation and optimization of
2. Brahmankar, D. M., and Sunil B.
physicochemical properties into drug discovery
Jaiswal. Biopharmaceutics
from target hits to final drug molecule can be
pharmacokinetics: A treatise. Vallabh
fruitful. During lead optimization and parameters
prakashan, 2005.
such as Absorption, Distribution, Metabolism,
3. Foye, William O. Foye's principles of
Excretion and Toxicity (ADME/Tox) properties
medicinal chemistry. Eds. Thomas L.
should be emphasized throughout the entire
Lemke, and David A. Williams. Lippincott
discovery process. This approach also helps to
Williams & Wilkins, 2008.
improve efficiency, as problematic compounds are
4. Kerns, Edward, and Li Di. Drug-like
removed and delay or failures of candidates are
properties: concepts, structure design and
optimization. Academic Press, 2010.
Article References:
"Determination of solute lipophilicity, as
log< i> P</i>(octanol) and log< i>
P</i>(alkane)
divinylbenzene) and immobilised artificial
membrane stationary phases in reversed-
high-performance
Figure 12. Flowchart for Optimized Drug
chromatography." Journal
Chromatography A 766.1 (1997): 35-47.
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
2. Ashwood, Valerie A., et al. "Utilization of
antiinflammatory
an intramolecular hydrogen bond to
activity." Journal
medicinal
increase the CNS penetration of an NK1
chemistry 47.18 (2004): 4517-4529.
receptor antagonist." Journal of medicinal
8. Cheng, Menyan, et al. "Design and
chemistry 44.14 (2001): 2276-2285.
piperazine-based
3. Avdeef, Alex. "Physicochemical profiling
metalloproteinase inhibitors." Journal of
(solubility, permeability
medicinal chemistry 43.3 (1999): 369-380.
state)." Current topics in medicinal
9. Clark, David E. "< i> In silico</i>
chemistry 1.4 (2001): 277-351.
4. Bighley, Philip L. "Salt selection for basic
permeation."Drug discovery today 8.20
International
(2003): 927-933.
Pharmaceutics 33.1 (1995): 201-217.
10. Comer, John, and Kin Tam. "Lipophilicity
profiles: theory and measurement."Testa,
Pharmacokinetics made easy. Sydney:
B.; van de Waterbeemd, H.; Folkers,
G (2001): 275-304.
6. Borgos, Sven EF, et al. "Probing the
11. Di, Li, and Edward H. Kerns. "Application
structure-function relationship of polyene
of physicochemical data to support lead
macrolides: engineered biosynthesis of
soluble nystatin analogues." Journal of
Optimizing
"drug-like"
medicinal chemistry 49.8 (2006): 2431-
properties of leads in drug discovery.
Springer New York, 2006. 167-193.
7. Chen, Ping, et al. "Imidazoquinoxaline
12. Di, Li, and Edward H. Kerns. Methods for
Src-family kinase p56Lck inhibitors: SAR,
Assessing
Blood–Brain
QSAR, and the discovery of (S)-N-(2-
Penetration in Drug Discovery. John
Wiley & Sons, New York, 2011.
piperazinyl) imidazo-[1, 5-a] pyrido [3, 2-
13. Di, Li, et al. "Evidence-based approach to
e] pyrazin-6-amine (BMS-279700) as a
assess passive diffusion and carrier-
potent and orally active inhibitor with
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
mediated drug transport." Drug discovery
19. Hann, Mike M., and Tudor I. Oprea.
today 17.15 (2012): 905-912.
"Pursuing the leadlikeness concept in
14. Di, Li, et al. "High throughput artificial
pharmaceutical research." Current opinion
membrane permeability assay for blood–
in chemical biology 8.3 (2004): 255-263.
brain barrier." European journal of
20. Hansch, Corwin, et al. "Qsar and
medicinal chemistry 38.3 (2003): 223-232.
Bioorganic
medicinal
chemistry12.12 (2004): 3391-3400.
Diarylthiophenes as selective EP< sub>
21. Johanson, Conrad E., et al. "Enhanced
1</sub> receptor antagonists." Bioorganic
prospects for drug delivery and brain
& medicinal chemistry letters 15.4 (2005):
targeting by the choroid plexus–CSF
route." Pharmaceutical
research 22.7
16. Ellens, Harma, et al. "In vitro permeability
(2005): 1011-1037.
screening for identification of orally
22. John Arrowsmith & Philip Miller; Trial
Watch: Phase II and Phase III attrition
antagonists." Advanced drug delivery
rates 2011–2012; Nature Reviews Drug
reviews23.1 (1997): 99-109.
Discovery 12, 569, (2013).
17. Goodwin, Jay T., and David E. Clark. "In
23. Kassel, Daniel B. "Applications of high-
silico predictions of blood-brain barrier
penetration: considerations to "keep in
discovery."Current opinion in chemical
mind"." Journal of Pharmacology and
biology 8.3 (2004): 339-345.
Experimental Therapeutics 315.2 (2005):
18. Goodwin, Jay T., et al. "Physicochemical
discovery. Drug discovery today, 8(7),
permeability: role of solute hydrogen-
25. Kerns, E. H., and L. Di. "Physicochemical
bonding potential and volume." Journal of
profiling: overview of the screens."Drug
medicinal chemistry 44.22 (2001): 3721-
Discovery Today: Technologies 1.4 (2004):
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
26. Kerns, Edward H. "High throughput
pharmacological
toxicological
methods, 44(1), 235-249.
discovery." Journal
pharmaceutical
sciences 90.11 (2001): 1838-1858.
properties and the causes of poor solubility
27. Lee, Yung-Chi, Philip D. Zocharski, and
permeability."
Brian Samas. "An intravenous formulation
pharmacological
toxicological
decision tree for discovery compound
methods44.1 (2000): 235-249.
33. Lipinski, Christopher A. "Lead-and drug-
journal of pharmaceutics 253.1 (2003):
revolution."
Discovery
28. Li, Ying, et al. "Artemisinin derivatives:
Technologies 1.4 (2004): 337-341.
activity." Bioorganic & medicinal chemistry
"Experimental
11.20 (2003): 4363-4368.
approaches to estimate solubility and
29. Lin, Jiunn H., and Anthony YH Lu. "Role
of pharmacokinetics and metabolism in
settings." Advanced
delivery reviews 64 (2012): 4-17.
development." Pharmacological
35. Lipper, ROBERT A. "How can we
reviews 49.4 (1997): 403-449.
optimize selection of drug development
30. Lin, Jiunn H., and Anthony YH Lu. "Role
candidates from many
of pharmacokinetics and metabolism in
36. Liu, Xingrong, et al. "Unbound drug
concentration in brain homogenate and
development." Pharmacological
cerebral spinal fluid at steady state as a
reviews 49.4 (1997): 403-449.
surrogate for unbound concentration in
31. Lipinski, C. A. (2000). Drug-like
brain interstitial fluid." Drug Metabolism
properties and the causes of poor solubility
and Disposition 37.4 (2006): 787-793.
and poor permeability. Journal of
37. Lobell, Mario, László Molnár, and György
M. Keserü. "Recent advances in the
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
prediction of blood–brain partitioning
from molecular structure." Journal of
41. Oprea, Tudor I. "Current trends in lead
pharmaceutical sciences 92.2 (2003): 360-
properties?."
38. Lombardo, Franco, et al. "Prediction of
computer-aided molecular design 16.5-6
volume of distribution values in humans
(2002): 325-334.
42. Pardridge, William M. "Crossing the
physicochemical measurements and plasma
blood–brain barrier: are we getting it
protein binding data." Journal of medicinal
right?." Drug discovery today 6.1 (2001):
chemistry 45.13 (2002): 2867- 2876.
43. Pardridge, William M. "Transport of small
39. Martin, Alfred N., James Swarbrick, and
molecules through the blood-brain barrier:
Arthur Cammarata. "Physical pharmacy:
biology and methodology." Advanced drug
delivery reviews 15.1 (1995): 5-36.
pharmaceutical sciences." (1993).
44. Pelkonen, R. Olavi, Andreas Baumann,
40. Maurer, Tristan S., et al. "Relationship
and Anhreas Reichel. Pharmacokinetic
between exposure and nonspecific binding
challenges in drug discovery. Vol. 37.
of thirty-three central nervous system drugs
Springer Verlag, 2002.
in mice." Drug metabolism and disposition
33.1 (2005): 175-181.
Mannhold. Calculation
Nikam, Sham S., et al. "Design and
lipophilicity: the hydrophobic fragmental
synthesis of novel quinoxaline-2, 3-dione
constant approach. Wiley-VCH, 1992.
amino acid derivatives." Journal of
medicinal
chemistry 42.12 (1999):
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
46. Roda, Aldo, et al. "Synthesis and
drug absorption and pharmacokinetics.
physicochemical,
Journal of Medicinal Chemistry, 44, 1313-
pharmacological properties of new bile
medicinal
chemistry 39.11 (1996): 2270-2276.
biopharmaceutics
47. Rowland, Malcolm, Carl Peck, and
(BCS): a commentary." Eur. J. Pharm. Sci
Geoffrey Tucker. "Physiologically-based
pharmacokinetics in drug development and
54. Veber, Daniel F., et al. "Molecular
science." Annual review of
pharmacology and toxicology 51 (2011):
candidates." Journal of medicinal chemistry
48. Rubin, L. L., and J. M. Staddon. "The cell
45.12 (2002): 2615-2623.
biology of the blood-brain barrier."Annual
55. Venkatesh, Srini, and Robert A. Lipper.
review of neuroscience 22.1 (1999): 11-28.
"Role of the development scientist in
49. Smith D. A. (2002). Ernst Schering
Research Foundation Workshop, 37, 203-
optimization." Journal of pharmaceutical
sciences89.2 (2000): 145-154.
50. Stella, Valentino J. "A case for prodrugs:
56. Wei, Zhong-Yong, et al. "N, N-Diethyl-4-
fosphenytoin." Advanced drug delivery
reviews 19.2 (1996): 311-330.
51. Stenberg, Patric, et al. "Theoretical
selective, potent δ opioid receptor agonist
predictions of drug absorption in drug
development."
analogues." Journal of
Medicinal
pharmacokinetics 41.11 (2002): 877-899.
chemistry 43.21 (2000): 3895-3905.
52. Van de Waterbeemd, H., Smith, D. A.,
57. William M. Pardridge, Transport of small
Beaumont, K., & Walker, D. K. (2001).
molecules through the blood-brain barrier:
Property Based Design: Optimisation of
biology and methodology, Advanced Drug
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
RA Journal of Applied Research
Volume 1 Issue 1 Pages-18-43 November-2014 ISSN (e): Appl
Delivery Reviews, Volume 15, Issues 1–3,
July 1995, Pages 5-36.
58. Wu, Chi-Yuan, and Leslie Z. Benet.
"Predicting drug disposition via application
and development of a biopharmaceutics
system." Pharmaceutical
research 22.1
59. Xie, Lan, et al. "Anti-AIDS
Agents." Journal
medicinal
Chemistry 42.14 (2004): 2662-2672.
60. Yalkowsky, Samuel Hyman, and Sujit
Banerjee. Aqueous solubility: Methods of
estimation for organic compounds. New
York: Marcel Dekker, 1992.
61. Zhang, Yuanchao, and Leslie Z. Benet.
"The gut as a barrier to drug absorption."
Clinical pharmacokinetics 40.3 (2001): 159-
Kapadia Akshay Bhupendra RAJAR Volume 1 Issue 1 November 2014
Source: http://rajournals.in/images/ijararticle/v1-i1/4ijar.Pdf
VK6N SERIES VIDEOKITS VIDEOKIT SERIE VK6N One Way and Two Way Norme 's Manual We recommendThis equipment is installed by aCompetent Electrician, Security orCommunications Engineer Factory - OfficeVIDEX ELECTRONICS S.p.A. Via del lavoro,1 63020 MONTEGIBERTO (AP) - ITALYPhone: (+39) 0734 - 631669 Fax: (+39) 0734 - 632475 www.videx.it e-mail: [email protected]
Ethio. J. Appl. Sci. Technol. Vol(4):65-75(2013) 65 ORIGINAL ARTICLE In vitro Nematicidal (Anthelmintic) Property of the Seed Extracts of Anamirta cocculus (Linn.) Against Pheretima posthuma (L. Vaill.) Umer Qadir and Paul, V.I.* Department of Zoology, Annamalai University, Annamalainagar 608 002, Tamil Nadu,