Web.yonsei.ac.kr
Supplemental Material can be found at:http://www.jbc.org/content/suppl/2012/04/05/M112.364281.DC1.html
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 287, NO. 28, pp. 23271–23282, July 6, 2012
2012 by The American Society for Biochemistry and Molecular Biology, Inc.
Published in the U.S.A.
Nigericin-induced Impairment of Autophagic Flux in
Neuronal Cells Is Inhibited by Overexpression of Bak*□
S
Received for publication, March 21, 2012, and in revised form, April 4, 2012 Published, JBC Papers in Press, April 5, 2012, DOI 10.1074/jbc.M112.364281
Junghyun Lim‡
, Yunsu Lee‡
, Hyun-Wook Kim§
, Im Joo Rhyu§
, Myung Sook Oh¶
, Moussa B. H. Youdim储
,
Zhenyu Yue**
, and Young J. Oh‡1
From the ‡
Department of Biology, Yonsei University College of Life Science and Biotechnology, 134 Shinchon-dong
Seodaemoon-gu, Seoul 120-749, Korea, the §
Department of Anatomy, College of Medicine, Korea University, Seoul 136-705, Korea,
the ¶
Department of Life and Nanopharmaceutical Science, Kyung Hee University, Seoul 130-701, Korea, 储
Technion Israel Institute of
Technology, Haifa 31096, Israel, and the **
Department of Neurology and Neuroscience, Friedman Brain Institute, Mount Sinai
School of Medicine, New York, New York 10029
Background: Exact mechanisms underlying regulatory roles for the Bcl-2 family remain to be delineated.
Results: Overexpressed Bak blocked ionophore-induced impairment of autophagic flux in neuronal cells.
Conclusion: Bak plays critical roles in maintaining autophagic flux and vacuole homeostasis.
Significance: Our findings contribute to better understanding of the Bcl-2 family's potential as a novel therapeutic strategy in
Bak is a prototypic pro-apoptotic Bcl-2 family protein
somal pathways (1). Among the three types of autophagy (
e.g.
expressed in a wide variety of tissues and cells. Recent studies
macroautophagy, microautophagy, and chaperone-mediated
have revealed that Bcl-2 family proteins regulate apoptosis as
autophagy), macroautophagy encompasses the formation of
well as autophagy. To investigate whether and how Bak exerts a
autophagosome and the subsequent autolysosome after fusion
regulatory role on autophagy-related events, we treated inde-
with the lysosome (2). Among several molecules involved in
pendent cell lines, including MN9D neuronal cells, with nigeri-
autophagy, LC3 has been widely used as a marker of autophagy.
cin, a Kⴙ
/Hⴙ
ionophore. Treatment of MN9D cells with nigeri-
Because soluble cytosolic LC3-I is modified through lipidation
cin led to an increase of LC3-II and p62 levels with concomitant
and predominantly associated with the autophagosomal mem-
activation of caspase. Ultrastructural examination revealed
brane, the levels of LC3-II that form in a given situation can be
accumulation of autophagic vacuoles and swollen vacuoles in
used to predict the extent of autophagosome formation (3). As
nigericin-treated cells. We further found that the LC3-II accu-
the LC3-II is degraded after fusion between autophagosomes
mulated as a consequence of impaired autophagic flux and the
and lysosomes, increased levels of LC3-II can result from either
disrupted degradation of LC3-II in nigericin-treated cells. In
induced synthesis of autophagosomes or a reduced rate of deg-
this cell death paradigm, both transient and stable overexpres-
radation (4). Maturation of autophagosomes is a multistep
sion of various forms of Bak exerted a protective role, whereas it
process involving fusion with early or late endosomes, such as
did not inhibit the extent of nigericin-mediated activation of
multivesicular bodies (MVBs),2 yielding the amphisome, prior
caspase-3. Subsequent biochemical and electron microscopic
to fusion with lysosomes (5, 6). Any defects in this process can
studies revealed that overexpressed Bak maintained autophagic
impair autophagic flux, leading to the accumulation of LC3-II.
flux and reduced the area occupied by swollen vacuoles in nige-
Functional MVBs or endosomal compartments are required for
ricin-treated cells. Similar results were obtained in nigericin-
efficient autophagosomal maturation. In some pathological
treated non-neuronal cells and another proton ionophore-in-
conditions, it has been reported that impaired MVBs or endo-
duced cell death paradigm. Taken together, our study indicates
somal proteins lead to abnormal autophagic degradation (7–9).
that a protective role for Bak during ionophore-induced cell
Cell death modality has been routinely classified as apopto-
death may be closely associated with its regulatory effect on
sis, necrosis, or autophagic cell death (10). Although there have
maintenance of autophagic flux and vacuole homeostasis.
been debates as to whether autophagy
per se executes cell deathor whether it represents a cytoprotective process (11, 12), thereis a growing body of evidence supporting the involvement of
Autophagy is an evolutionarily conserved catabolic process
bona fide autophagic cell death in various forms of pathophys-
involved in cellular homeostasis and stress responses, including
iological situations (13). Intriguingly, these distinct forms of cell
the degradation of target proteins or organelles through lyso-
death do not seem to occur in a mutually exclusive way, butrather cell death can be accompanied by mixed features of mul-tiple cell death mechanisms at the same time or sequentially
* This work was supported by grants from the Ministry of Science and Tech-
nology through BRC, and in part by the Korea Science and Engineering
(14, 15). Furthermore, the interrelationship between autophagy
Foundation (SRC, 2012-0000496) and the Mid-Career Research Programthrough NRF funded by the Ministry of Education, Science, and Technol-ogy (to Y. J. O.).
2 The abbreviations used are: MVB, multivesicular body; MEF, mouse embryo
This article contains
fibroblast; Z, benzyloxycarbonyl; AV, autophagic vacuole; MTT, 3-(4,5-di-
1 To whom correspondence should be addressed. Tel.: 82-2-2123-2662; Fax:
methylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; BH3 domain, Bcl-2
82-2-312-5657; E-mail:
[email protected].
homology domain 3.
JULY 6, 2012 • VOLUME 287 • NUMBER 28
JOURNAL OF BIOLOGICAL CHEMISTRY
Protective Role for Bak in Nigericin-induced Cell Death
and apoptosis has recently attracted much attention (13, 15).
time periods. Other drugs used in the study included 1 M stau-
These two distinct cell death modalities can act in a cooperative
rosporine, 0.625 M monensin, 10 M chloroquine, and 10 mM
manner to invoke cell death or in a disparate manner to antag-
ammonium chloride (all from Sigma), or 100 M Z-VAD-fluo-
onize each other. It has been indicated that the two processes
romethyl ketone (Enzyme System Products, Livermore, CA).
share common response machineries, including the B-cell lym-
All drug concentrations were empirically determined.
phoma 2 (Bcl-2) family, reactive oxygen species, and intracellu-
Construction of Eukaryotic Vectors and Transfection—Hu-
lar calcium (15). Bcl-2 family proteins are classified as either
man full-length Bak was provided by Dr. J. C. Reed (Burnham
anti-apoptotic or pro-apoptotic (16). For example, pro-apop-
Institute, La Jolla, CA). For construction of Myc-tagged Bak or
totic members of the Bcl-2 family accelerate apoptosis by
FLAG-tagged Bak constructs, N-terminal Myc or FLAG tags
antagonizing anti-apoptotic Bcl-2 proteins in several distinct
were added to full-length human Bak cDNA by polymerase
ways (17). Typically, two major pro-apoptotic proteins, Bcl-2
chain reaction (PCR; for Myc tag, 5⬘-GCC GGA TCC GCC
antagonist killer 1 (Bak) and Bcl-2-associated x protein (Bax),
ACC ATG GAA CAA AAG TTG ATT TCA GAA GAA GAT
accelerate mitochondrial outer membrane permeabilization via
CTG GCC ATG GCA TCT GGA CAA GGA CCA G-3⬘; for
conformational changes and subsequent oligomerization on
FLAG tag, 5⬘- GCC GGA TCC GCC ACC ATG GAC TAT
the mitochondrial membrane (18). The involvement of Bcl-2
AAG GAC GAT GAT GAC AAG GCC ATG GCA TCT GGA
family members or their interacting proteins in autophagy has
CAA GGA CCA G-3⬘; for all, 3⬘-CGG AAT TCT CAT GAT
recently been suggested because mammalian Beclin 1 has been
CTG AAG AAT CTG TGT AC-5⬘). PCR products were
demonstrated to be required for autophagy. Beclin 1 was ini-
digested with BamHI and EcoRI and ligated into the
tially identified as a Bcl-2-interacting protein, and its Bcl-2
pcDNA3.1(⫹) vector. GFP-tagged LC3 cDNA was provided by
homology domain 3 (BH3 domain) mediates interaction with
Dr. Yoshimory (Osaka University). Transfection was per-
Bcl-2 or Bcl-xL (19, 20). Similarly, it has been demonstrated
formed using Lipofectamine 2000 (Invitrogen), as recom-
that Bcl-2 family proteins influence autophagy mainly by regu-
mended by the supplier. The parent vector was used as a
lating the interaction between Beclin 1 and Bcl-2/Bcl-xL (20 –
control. Stable cell lines overexpressing Myc-tagged (MN9D/
24). Nevertheless, the exact mechanisms underlying the regu-
m-Bak) or FLAG-tagged Bak (MN9D/f-Bak) were also estab-
latory roles for Bcl-2 family members still remain to be further
lished and characterized, as described previously (31). The sta-
ble cell lines were cultured in DMEM supplemented with 10%
Nigericin is an ionophore and acts as an antiporter of K⫹/H⫹
FBS and 500 g/ml G-418 (A.G. Scientific, San Diego, CA). For
and raises the pH of acidic compartments (25). In image-based
gene silencing studies, two different pairs of siRNAs for human
screens for autophagy inducers, nigericin was identified as one
Bak (1011326 and 1011323 duplex) and negative siRNA (SN-
of the candidates that increase LC3 spots following treatment
1003) were purchased from Bioneer (Daejeon, Korea) and used
(26). In addition, it has been reported that nigericin treatment
for transfection. Each siRNA (50 nM) was transfected into the
reduces lysosomal protein degradation and inhibits fusion
indicated cells using Lipofectamine 2000.
between autophagosomes and lysosomes by raising the pH of
Electron Microscopy—As described previously in our labora-
acidic compartments (27, 28). In our previous study using this
tory (32), electron microscopic examination followed by quan-
ionophore, we demonstrated that Bax attenuates nigericin-in-
titative analysis was performed after MN9D/neo, or MN9D/f-
duced MN9D dopaminergic neuronal cell death, although the
Bak cells were incubated with or without 0.1 M nigericin.
exact mechanisms behind Bax-mediated regulation of cell
Briefly, ultrastructural examination of 80-nm thin sections was
death were not elucidated (29). Here, we primarily used MN9D
performed on a Hitachi H-7500 transmission electron micro-
dopaminergic neuronal cells to investigate (i) whether nigeri-
scope with 80-kV acceleration voltage. Quantitative analysis of
cin-induced neurodegeneration is accompanied by typical
the electron photomicrographs for the average numbers of
morphological and biochemical features of autophagy and (ii)
autophagic vacuoles (AVs) per cell and areas occupied by swol-
whether and how Bak regulates nigericin-induced autophagic
len vacuoles was conducted using Scion Image software (Scion
processes. In sum, we found that Bak attenuated nigericin-in-
Corp.). All quantitative analyses were calculated from 30 ran-
duced cell death potentially via its regulation of autophagic flux
domly selected cells in two independent experiments.
during ionophore-induced cell death.
Fluorescence and Phase-contrast Microscopy—After incuba-
tion with nigericin for the indicated time periods, cells were
fixed with 4% paraformaldehyde (EMS, Hatfield, PA) for 15 min
Cell Culture and Drug Treatment—MN9D cells, N18TG
and then permeabilized with 0.1% saponin (Sigma) for 10 min at
cells, and HEK293 cells were cultured in Dulbecco's modified
room temperature. Cells were then blocked in phosphate-buff-
Eagle's medium (DMEM; Sigma) supplemented with 10% heat-
ered saline (PBS; Lonza, Basel, Switzerland) containing 0.2%
inactivated fetal bovine serum (FBS) from Invitrogen. Culture
Triton X-100 and 5% normal goat serum (Invitrogen). Cells
medium was switched to serum-free N2 medium for drug treat-
were then incubated with rabbit anti-LC3 antibody (1:200; Cell
ment as described previously (30). Atg5 wild-type (WT) and
Signaling, Beverly, MA) or mouse anti-Lamp1 antibody (1:200;
knock-out (KO) MEF cells were cultured in DMEM supple-
Developmental Studies Hybridoma Bank, Iowa City, IA) in PBS
mented with 10% FBS and 1% penicillin-streptomycin (Invitro-
containing 0.2% Triton X-100 and 1% normal goat serum over-
gen). Cells were exposed to predetermined concentrations of
night at 4 °C. After extensive washing with PBS, cells were incu-
nigericin (Sigma; 0.1, 0.5, 1, or 2.5 M nigericin for MN9D,
bated with Alexa 488-conjugated goat anti-rabbit IgG or Alexa
N18TG, HEK293, or MEF cells, respectively) for the indicated
488-conjugated goat anti-mouse IgG (1:200; all from Invitro-
23272 JOURNAL OF BIOLOGICAL CHEMISTRY
VOLUME 287 • NUMBER 28 • JULY 6, 2012
Protective Role for Bak in Nigericin-induced Cell Death
gen) at room temperature for 1 h. Nuclei were stained with
of cells in each quadrant over the total number of the counted
Hoechst 33258 (1 g/ml; Molecular Probes, Inc., Eugene, OR)
cells was calculated using CellQuest (BD Biosciences). Follow-
for 10 min at room temperature, and slides were mounted in
ing drug treatment, the rate of cell viability was measured by the
Vectashield mounting medium (Vector Laboratories, Burlin-
game, CA). Images were acquired under an LSM 510 META
(MTT) reduction assay, as described previously (33). Phase-
confocal laser-scanning microscope equipped with epifluores-
contrast photomicrographs were taken with an Axio Observer
cence and a digital image analyzer (Carl Zeiss, Zena, Germany).
A1 microscope (Carl Zeiss).
As previously described by us (32), acquired images were ana-
Statistics—Data are presented as the mean ⫾ S.D. from three
lyzed for LC3 spots using MetaMorph (Molecular Devices,
independent experiments, unless otherwise indicated. Stu-
Downingtown, PA). Each Z series was maximum-projected and
dent's
t tests were used for determining significant differences
then used for quantitation of areas and numbers of LC3 spots
between groups. Values of
p ⬍ 0.001 or
p ⬍ 0.05 were consid-
per cell. Thirty randomly selected cells from three independent
ered statistically significant.
experiments were subjected to quantitative analysis. To obtainlive images of GFP-tagged LC3, cells treated with or without 0.1
M nigericin for the indicated time periods were washed with
Characterization of Nigericin-induced MN9D Neuronal Cell
PBS and photographed under an Axio Observer A1 microscope
Death—To unequivocally characterize and quantify nigericin-
(Carl Zeiss).
induced cell death, we performed multiple, methodogically
Immunoblot Analysis—Following drug treatment, MN9D,
unrelated assays, as previously described (10). First, we exam-
N18TG, HEK293, or MEF cells were lysed with radioimmune
ined the morphological changes of MN9D cells treated with or
precipitation buffer and processed as described previously (32).
without nigericin for 12 h under a phase-contrast microscope.
The primary antibodies used included rabbit anti-LC3 (1:4,000;
As shown in Fig. 1
A, nigericin induced the dramatic accumula-
Cell Signaling), guinea pig anti-p62 (1:5,000; Progen Biotech-
tion of vacuole-like structures in the cytosol. As determined by
nik, Heidelberg, Germany), rabbit anti-cleaved caspase-3
MTT reduction assays, the viability of MN9D cells decreased
(1:1,000; Cell signaling), mouse anti-Myc antibody (1:2,000;
following nigericin treatment in a time-dependent manner (Fig.
Cell Signaling), rabbit anti-Bak (1:1,000; Upstate, Lake Placid,
1
B). Quite similar patterns were obtained by a colorimetric
NY), rabbit anti-GFP (1:1,000; Santa Cruz Biotechnology, Inc.,
measurement using a well known vital staining dye, neural red
Santa Cruz, CA), rabbit anti-Atg5 (1:3,000; Abcam, Cambridge,
(71.2 ⫾ 6.8% at 12 h, and 39.3 ⫾ 10.6% at 18 h over the untreated
MA), and HRP-conjugated anti-FLAG (1:5,000; Sigma). Dupli-
control cells;
n ⫽ 3,
p ⬍ 0.05). Next, we monitored double
cate blots probed with mouse anti-GAPDH (1:4,000; Chemi-
staining patterns of FITC-conjugated Annexin V and pro-
con, Dundee, UK) or rabbit anti-actin (1:3,000; Sigma) were
pidium iodide by FACS. Our FACS analysis revealed that nige-
used as loading controls. After incubation with primary anti-
ricin-induced cell death produced a mixture of population pos-
body, membranes were washed with Tris-buffered saline con-
itive for Annexin V alone and population positive for both
taining 0.1% Tween 20, followed by incubation with the appro-
Annexin V and propidium iodide (Fig. 1
C). Analyses with Mito-
priate horseradish peroxidase-conjugated anti-rabbit (1:4,000;
tracker Orange indicated a collapse of mitochondrial potential
Santa Cruz Biotechnology, Inc.), anti-mouse (1:4,000; Santa
(⌬⌿) following nigericin treatment (Fig. 1
D). Immunoblot
Cruz Biotechnology, Inc.), or anti-guinea pig antibody (1:4,000;
analyses using antibodies recognizing a cleaved caspase-3
Sigma). Specific bands were visualized using the enhanced
revealed that caspase-3 activity increased following nigericin
chemiluminescence kit (PerkinElmer Life Sciences). After
treatment (Fig. 1
E). Co-treatment of cells with Z-VAD-fluo-
measuring the intensity of each band by densitometry using
romethyl ketone (a pan-caspase inhibitor) blocked nigericin-
ImageJ, relative intensities were calculated by normalizing to
induced cell death (Fig. 1
F). Similarly, a neutral red-based col-
GAPDH from the corresponding sample.
orimetric assay demonstrated that a caspase inhibitor
Activity and Viability Assays—Following nigericin treat-
attenuates drug-induced cell death (at 18 h, 39.3 ⫾ 10.6% for
ment, MN9D cells were lysed with the InnoZymeTM cathepsin
nigericin alone
versus 67.2 ⫾ 9.4% for nigericin plus Z-VAD;
L activity kit (Calbiochem), as recommended by the supplier.
n ⫽ 3,
p ⬍ 0.05).
Cell lysates were then incubated with fluorogenic substrate
Characterization of Autophagic Events following Nigericin
Z-Phe-Arg-7-amido-4-methylcoumarin for 1 h at 37 °C. For
Treatment—Based on previous reports by others (26 –28), we
measurement of mitochondrial membrane potential, cells were
hypothesized that nigericin-induced cell death may also recruit
incubated with 250 nM Mitotracker Orange CMXRos (Invitro-
autophagic events. To directly test this, we first examined ultra-
gen) for 30 min at 37 °C and then washed twice with PBS. Flu-
structural changes in MN9D cells before and after nigericin
orescence intensity was measured at Ex360/Em460 nm (excita-
treatment. Based on the previous report describing in detail
tion/emission wave length for cathepsin activity) or at Ex554/
ultrastructural criteria and characterization to monitor
Em576 nm (for Mitotracker Orange) using a FL600 microplate
autophagy (34), we identified four important structures:
reader (BioTek, Winooski, VT). Nigericin-treated MN9D cells
autophagosome, autolysosome, swollen endosome, and MVB.
were subjected to FACS analysis of Annexin V binding and
We found that MN9D cells exposed to nigericin for 7 h under-
propidium iodide uptake using the FITC Annexin V Apoptosis
went dramatic morphological changes (Fig. 2
A,
top panels).
Detection Kit 1 (BD Biosciences). Cells were stained with pro-
The presence of AVs that are typical of autophagy was found
pidium iodide/Annexin V as per the supplier's instructions and
within the cytosol (Fig. 2
A,
bottom left panels). Quantitative
analyzed on a FACSCalibur (BD Biosciences). The percentage
analyses indicated that the numbers of AVs increased following
JULY 6, 2012 • VOLUME 287 • NUMBER 28
JOURNAL OF BIOLOGICAL CHEMISTRY
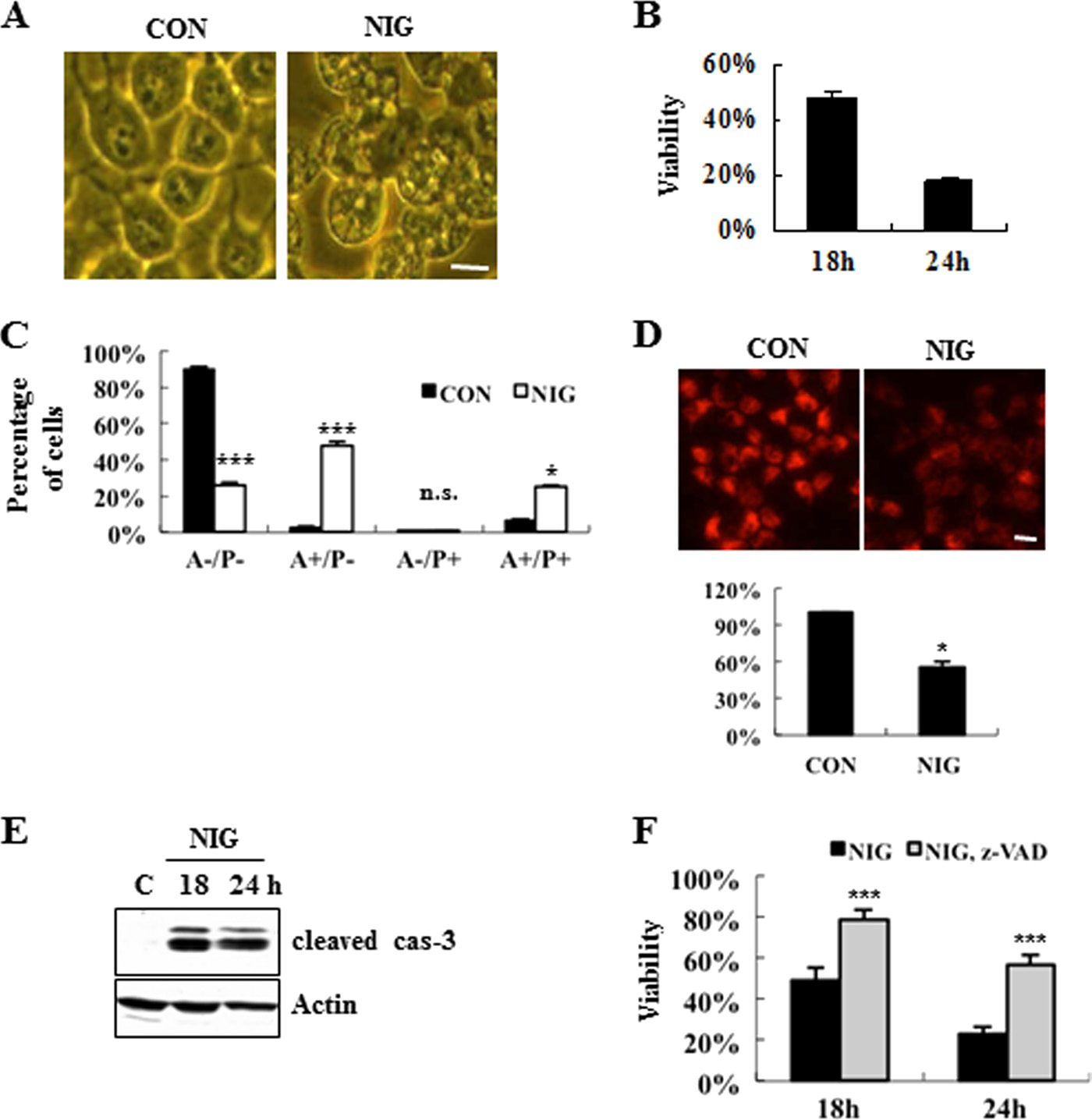
Protective Role for Bak in Nigericin-induced Cell Death
FIGURE 1. Characterization of nigericin-induced MN9D neuronal cell death. Multiple assays were performed to quantify nigericin-induced neuronal cell
death as described previously (10). A, phase-contrast images of MN9D cells incubated for 12 h with 0.1 M nigericin (NIG) or without drug (CON). Scale bar, 10
m. B, MTT reduction assays were performed following 0.1 M nigericin treatment for the indicated time periods. Viability after each treatment was expressedas a percentage over the untreated control cells (100%). Each point represents the mean ⫾ S.D. (error bars) from three independent experiments done intriplicate. C, FACS analysis on Annexin V (A) and propidium iodide (P) was performed following treatment with 0.1 M nigericin for 18 h. Values from FACSanalysis were shown as a percentage of the cell numbers from each quadrant over the total number of the counted cells from three independent experiments.
D, to measure the mitochondrial membrane potential, MN9D cells were loaded with 250 nM Mitotracker Orange CMXRos after incubation with or without 0.1
M nigericin. Fluorescence photomicrographs were taken at 18 h after nigericin treatment under a confocal microscope. Scale bar, 10 m. The relative intensitywas quantified using a microplate reader and expressed as a percentage of the untreated control cells (100%). Bar, mean ⫾ S.D. from three independentexperiments in triplicate. E, following nigericin treatment, caspase-3 activities were assessed by immunoblot analysis using anti-cleaved caspase-3. Actin wasused as a loading control. F, viability was measured by the MTT reduction assay after exposure to 0.1 M nigericin alone or in combination with 100 MZ-VAD-fluoromethyl ketone for the time periods indicated. Viability was expressed as a percentage over the untreated control cells (100%). Bar, mean ⫾ S.D.
from three independent experiments in triplicate. *, p ⬍ 0.05; ***, p ⬍ 0.001; n.s., not significant.
nigericin treatment (Fig. 2A, bottom right panel). In addition,
(right panel). Quantitative analyses demonstrated that the total
the appearance of swollen vacuoles (black arrows) was detected,
area of LC3 spots as well as the numbers of LC3 punctated spots
which is presumably related to endosomal systems (35). Next,
increased after nigericin treatment for 12 h (Fig. 2D). Similarly,
we attempted to check the involvement of autophagy during
we observed a punctated staining pattern for exogenous GFP-
nigericin-induced cell death on a biochemical level, and we per-
tagged LC3 from live cell fluorescence images (Fig. 2E) and an
formed immunoblot analysis using an antibody that recognizes
accumulation of GFP-LC3-II (Fig. 2F) in nigericin-treated
LC3. Fig. 2B shows that nigericin treatment induced accumu-
MN9D cells stably transfected with a eukaryotic vector con-
lation of the endogenous LC3-II form. Due to localization of
taining GFP-tagged LC3 cDNA. Collectively, our data demon-
LC3-II at the autophagosomal vesicles, the formation and
strated that both apoptotic and autophagic events were induced
punctated staining pattern of LC3-II forms are also widely used
during nigericin-mediated cell death. As shown in
to monitor autophagy. To effectively visualize its punctated
however, co-treatment with Z-VAD did not affect
staining pattern, we used saponin as a permeabilizing reagent
the nigericin-induced levels of LC3-II, implying that nigericin-
that removes soluble LC3 proteins from the cells and efficiently
induced apoptotic signal may not be linked to autophagic
leaves the membrane-associated form of LC3 proteins within
the cytosol. As shown in Fig. 2C, immunocytochemical local-
Impaired Autophagic Flux following Nigericin Treatment—
ization analysis revealed that a prominently punctated staining
Using lysomotropic agents, such as chloroquine and ammo-
pattern for LC3 was detected in cells treated with nigericin
nium chloride, that inhibit protein degradation by raising
23274 JOURNAL OF BIOLOGICAL CHEMISTRY
VOLUME 287 • NUMBER 28 • JULY 6, 2012

Protective Role for Bak in Nigericin-induced Cell Death
FIGURE 2. Ultrastructural and biochemical characterization of nigericin-induced MN9D neuronal cell death. A, representative electron micrographs of
MN9D cells treated for 7 h with (NIG) or without (CON) 0.1 M nigericin. Black scale bars, 5 m. Swollen vacuoles in nigericin-treated cell are indicated by black
arrows. Ultrastructural identification of AVs (autophagosome and autolysosome) appeared in MN9D cells was basically made by the guideline provided in the
previous work (34). Enlarged images of typical autophagosome (a), autolysosome (b), MVB (c), and swollen endosome (d) frequently found in nigericin-treated
cells are shown in the left, bottom panels. White scale bars, 250 nm. The average numbers of AVs per cell were quantified from 30 randomly selected cells from
two independent experiments. Bars, average numbers of AVs per cell. B, levels of LC3-II were assessed by immunoblot analysis using anti-LC3 antibody after
incubation with or without 0.1 M nigericin. GAPDH was used as a loading control. Densitometric analysis was performed using ImageJ. The relative intensity
of LC3-II in each sample was calculated after normalization to GAPDH in the corresponding sample. Each bar represents the mean ⫾ S.D. (error bars) from three
independent experiments. C, confocal immunofluorescence images after immunocytochemistry using anti-LC3 antibody in cells treated for 12 h with or
without 0.1 M nigericin. Saponin was used to permeabilize the cells to remove the soluble proteins in the cytosol. Nuclei were stained with 1 g/ml Hoechst
33258. Scale bar, 10 m. D, average area and number of LC3 spots per cell were quantified as described under "Experimental Procedures." Thirty cells from each
of three independent experiments were quantified. E, representative live images from MN9D cells stably expressing GFP-LC3 before and after nigericin
treatment for 12 h. Scale bar, 10 m. F, the level of GFP-LC3-II was assessed through immunoblot analysis using anti-GFP antibody. GAPDH was utilized as a
loading control. The relative intensity of LC3-II in each sample was calculated after normalization to GAPDH in the corresponding sample. Each bar represents
the mean ⫾ S.D. from three independent experiments. *, p ⬍ 0.05; ***, p ⬍ 0.001.
intralysosomal pH (36), we specifically investigated whether the
p62/SQSTM1 over 18 h, supporting the notion that nigericin
nigericin-induced accumulation of LC3-II is due to enhanced
impaired the autophagic degradation process (Fig. 3B). To
synthesis or reduced degradation. When we compared and
determine which step may be responsible for the compromised
quantitated the levels of LC3-II in nigericin-treated cells in the
autophagy, we monitored the total cathepsin activity in an in
presence or absence of chloroquine or ammonium chloride, we
vitro cathepsin substrate assay. As shown in Fig. 3C, nigericin
found that co-treatment with lysomotropic agents did not lead
caused a reduction of cathepsin activity in MN9D cells. More-
to further increase in nigericin-induced accumulation of LC3-II
over, immunocytochemical localization studies showed that
(Fig. 3A), suggesting that accumulation of LC3-II was most
the signal from Lamp1 was diminished following nigericin
likely caused by an impairment of autophagic flux. p62/
treatment compared with control cells (Fig. 3D), implying that
SQSTM1 is widely used to monitor autophagy, due to its local-
nigericin may lead to lysosomal dysfunction. To compare via-
ization at the autophagic compartments and subsequent deg-
bility and expression patterns of LC3-II and p62, we also uti-
radation through autophagy (37). Immunoblot analysis
lized Atg5 WT and KO MEF cells (Fig. 4A). We found that
demonstrated that nigericin treatment increased the level of
nigericin-induced cell death was accelerated in Atg5 KO MEF
JULY 6, 2012 • VOLUME 287 • NUMBER 28
JOURNAL OF BIOLOGICAL CHEMISTRY


Protective Role for Bak in Nigericin-induced Cell Death
FIGURE 3. Impaired autophagic flux during nigericin-induced MN9D neuronal cell death. A, autophagic flux was monitored in cell lysates exposed to 0.1
M nigericin for the time periods indicated in the presence or the absence of 10 M chloroquine (CQ) or 10 mM ammonium chloride (NH Cl). Relative intensity
of LC3-II normalized to GAPDH was calculated. Each bar represents the mean ⫾ S.D. (error bars) from three independent experiments. B, following treatmentwith 0.1 M nigericin for the indicated time periods, immunoblot analysis was performed to detect protein levels of p62. GAPDH was used as a loading control.
An asterisk indicates modified forms of p62. C, in vitro cathepsin activity assays were performed, as described under "Experimental Procedures." Values areexpressed as a percentage of the untreated control cells (100%). Each bar represents the mean ⫾ S.D. from three independent experiments. D, MN9D cellstreated with or without 0.1
M nigericin for 18 h were subjected to immunocytochemical analysis using anti-Lamp1 antibody. Representative confocal images
are provided. Scale bar, 10 m. *, p ⬍ 0.05; n.s., not significant.
cells compared with that in Atg5 WT MEF cells (Fig. 4B). Asexpected, LC3-II was hardly seen, and an increased level of p62was found in Atg5 KO MEF cells (Fig. 4C). Taken together, ourdata suggest that autophagy is associated with nigericin-in-duced cell death, and, most importantly, impairment ofautophagic flux presumably through lysosomal dysfunction isresponsible for nigericin-induced cell death.
Bak-mediated Attenuation of Nigericin-induced Cell Death—
Based on our previous studies, demonstrating a dual role for thepro-apoptotic Bcl-2 family in protecting cells from nigericin-induced cell death while accelerating staurosporine-inducedcell death (29), we tested whether and how Bak exerts a protec-tive role in these cell death paradigms. First, we conductedMTT reduction assays in staurosporine- or nigericin-treatedMN9D cells, transiently or stably transfected with a vectorencoding human Bak cDNA or empty vector as a control.
When MN9D cells transiently or stably transfected with Bakwere exposed to staurosporine for 18 h, they were more vul-nerable to the cell death-inducing stimuli In contrast, MN9D cells transiently overexpressing Bakwere ⬃20% more resistant to cell death following treatmentwith nigericin (Fig. 5, A and B). This protective effect of Bakwas consistently reproduced in nigericin-treated MN9D
FIGURE 4. Effect of Atg5 knock-out on the rate of cell viability and levels
of LC3-II and p62. A, cell lysates from Atg5 WT MEF and Atg5 KO MEF cells
cells stably overexpressing Myc-tagged or FLAG-tagged Bak
were probed with anti-Atg5 antibody. Actin was used as a loading control. B,
(Fig. 5, C and D). To determine if the protective role for Bak
MTT reduction assay was carried out following 2.5 M nigericin treatment for24 h. Viability is expressed as a percentage over the untreated control cells
was cell type-specific, N18TG neuroglioma cells stably
(100%) from three independent experiments in triplicate. C, immunoblot
transfected with or without Bak were exposed to nigericin
analysis was then performed using anti-LC3, -p62, and -actin antibodies. Rel-
and subjected to the MTT reduction assays. As shown in Fig.
ative intensity of LC3-II in each condition was calculated and compared, asdescribed above. *, p ⬍ 0.05. Error bars, S.D.
5, E and F, two independent lines overexpressing Bak signif-
23276 JOURNAL OF BIOLOGICAL CHEMISTRY
VOLUME 287 • NUMBER 28 • JULY 6, 2012

Protective Role for Bak in Nigericin-induced Cell Death
quent quantitative analysis (Fig. 6D). Furthermore, nigericin-induced accumulation of p62 was inhibited in Bak-overexpress-ing cells (Fig. 6, C and D). To confirm that this effect isgenuinely caused by Bak, we established human embryonic kid-ney HEK293 and MN9D cells transfected with control siRNAor siRNA targeting human Bak mRNA. When we first com-pared the relative intensities of LC3-II over GAPDH in HEK293cells treated with or without nigiricin, we found that knock-down of endogenous Bak increased the level of LC3-II, whereasoverexpression of human Bak diminished the relative intensityof LC3-II (Fig. 7, A and B). Because MN9D cells express quitelow levels of endogenous Bak, we first established MN9D cellstransiently overexpressing FLAG-tagged Bak and then targetedthe exogenous Bak via two independent Bak siRNAs (#1 and #2in Fig. 7C). As shown in Fig. 7C, the levels of LC3-II in BaksiRNA-transfected MN9D cells were significantly increasedcompared with negative siRNA-transfected MN9D cells fol-lowing nigericin treatment. Taken together, our data indicate
that Bak attenuated the nigericin-induced accumulation ofLC3-II in both neuronal and non-neuronal cells and that itsinhibitory effect seemed to be independent of caspase activity.
Bak Attenuates Nigericin-induced Cell Death through Regu-
FIGURE 5. Attenuation of nigericin-induced cell death by overexpressed
lating Autophagic Flux—Next, we examined whether the Bak-
Bak. After transient transfection of MN9D cells with an expression vector
mediated reduction of LC3-II was a consequence of inhibiting
encoding human Bak (A), stable transfection of MN9D cells with empty vector
autophagy induction or of accelerating autophagic flux.
(n) and a vector encoding either FLAG-tagged human Bak (f) or Myc-taggedBak (m) (C), and stable transfection of N18TG cells with empty vector (n) or
Toward this end, we treated MN9D/neo and MN9D/f-Bak cells
human Bak (E), protein levels were detected by immunoblot analysis using
with nigericin in the presence or absence of chloriquine and
anti-Bak antibody. GAPDH or actin was used as a loading control. B, D, and F,MTT reduction assay was carried out following 0.1 M nigericin treatment for
subsequently performed immunoblot analysis to measure the
18 and 24 h (B) or 24 h (D and F). Viability after each treatment is expressed as
levels of LC3-II. Exposure of MN9D/f-Bak cells to nigericin in
a percentage over the untreated control cells (100%) from three independent
the presence of chloroquine tremendously increased the levels
experiments in triplicate. *, p ⬍ 0.05. Error bars, S.D.
of LC3-II, compared with MN9D/f-Bak cells treated with nige-
icantly protected N18TG cells from nigericin-induced
ricin alone (Fig. 8A). Although treatment of chloroquine alone
only slightly increased the basal level of LC3-II (refer to Fig. 3A),
Inhibitory Role of Bak in Nigericin-mediated Accumulation of
co-treatment with nigericin significantly increased LC3-II,
LC3-II—To determine the underlying protective mechanism
indicating a synergic effect of chloroquine in MN9D/f-Bak
exerted by Bak, we compared the levels of the cleaved form of
cells. Furthermore, the relative intensity of nigericin-induced
caspase-3 and LC3-II in nigericin-treated or non-treated
LC3-II was similar in both MN9D/neo and MN9D/f-Bak at
MN9D cells stably overexpressing control empty vector or
18 h, in the presence of chloroquine. This phenomenon was
FLAG-tagged or Myc-tagged Bak (MN9D/neo, MN9D/f-Bak,
reproduced when we treated cells with nigericin in combina-
or MN9D/m-Bak, respectively). Immunoblot analyses of these
tion with another lysomotropic agent, ammonium chloride
stable cell lines showed that nigericin-induced caspase activity
(data not shown). We next investigated whether Bak is critically
was similar or slightly increased in two independent stable cell
involved in the maintenance of autophagic flux during cell
lines overexpressing Bak (Fig. 6A). In contrast, there was a sig-
death induced by agents that have properties similar to those of
nificant difference in the levels of LC3-II between nigericin-
nigericin. The structure and properties of nigericin are similar
treated MN9D/neo and the two MN9D/Bak stable cell lines.
to those of monensin, although monensin has a preference for
Quantitative analysis revealed that the relative intensities of
Na⫹ instead of K⫹ (25, 38). Like nigericin, monensin treatment
LC3-II over GAPDH were significantly diminished in the two
disrupts lysosomal function (27, 39). Immunoblotting with
independent MN9D/Bak cell lines after treatment with nigeri-
anti-LC3 antibody and quantitative analysis showed that the
cin for 12 and 18 h (Fig. 6A, bottom panel). In support of this
time-dependent increase in LC3-II following monensin treat-
finding, in an immunocytochemical localization study, reduced
ment was not exacerbated by the presence of chloroquine, indi-
levels of LC3 spots were detected in MN9D/f-Bak cells follow-
cating that monensin also impaired autophagic flux in MN9D
ing nigericin treatment (Fig. 6B, top panels). Quantitative anal-
cells MTT reduction assays
yses demonstrated a significant reduction in the areas occupied
indicated that monensin-induced cell death was attenuated in
by LC3 spots and the total numbers of punctated LC3 spots per
MN9D cells stably overexpressing Bak
cell in nigericin-treated MN9D/f-Bak cells (Fig. 6B, bottom
In these MN9D/m-Bak cells, the rate of monensin-induced
panels). This phenomenon was reproduced when MN9D cells
accumulation of LC3-II was further increased in the presence of
transiently transfected with Bak were exposed to nigericin and
chloroquine As shown in
subjected to immunoblot analysis of LC3 (Fig. 6C) and subse-
the relative intensity of monensin-induced LC3-II
JULY 6, 2012 • VOLUME 287 • NUMBER 28
JOURNAL OF BIOLOGICAL CHEMISTRY

Protective Role for Bak in Nigericin-induced Cell Death
FIGURE 6. Bak hinders nigericin-induced accumulation of LC3-II. A, representative blots of stable cell lines overexpressing an empty vector (MN9D/neo),
FLAG-tagged Bak (MN9D/f-Bak), or Myc-tagged Bak (MN9D/m-Bak) were shown. Following exposure to 0.1 M nigericin for the indicated time periods, cell
lysates were harvested, transblotted, and probed with antibodies that recognize the cleaved caspase-3, LC3, and GAPDH, respectively. GAPDH was used as a
loading control. Relative intensity of LC3-II normalized to GAPDH was calculated after densitometric analysis using ImageJ (bottom). B, using anti-LC3 antibody,
confocal immunofluorescence images of MN9D/neo and MN9D/f-Bak were obtained after treatment with or without 0.1 M nigericin for 12 h. Scale bar, 10 m.
The average area and number of LC3 spots per cell were measured using Metamorph and determined as described in the legend to Fig. 2C. C, MN9D cells were
transiently transfected with empty vector (⫺) or human Bak (⫹) and then exposed to 0.1 M nigericin for 12 h. Subsequently, cells were lysed and probed with
anti-Bak, -LC3, -p62, and -GAPDH antibodies. An asterisk indicates modified forms of p62. D, relative intensity of LC3-II and p62 in each cell line was calculated
and compared, as described above. A, B, and D, all values for quantitative analyses represent the mean ⫾ S.D. (error bars) from three independent experiments.
*, p ⬍ 0.05; n.s., not significant.
was also similar in both MN9D/neo and MN9D/m-Bak cells at
(35), the appearance of these swollen vacuoles seemed to be
18 h, in the presence of chloroquine. Collectively, these results
related to endosomal systems. On the other hand, swollen vac-
support the notion that Bak seems to attenuate carboxylic iono-
uoles were less frequently observed in nigericin-treated
phore-induced cell death by maintaining autophagic flux.
MN9D/f-Bak cells. Quantitative analysis of the electron micro-
Interestingly, we found that the rate of nigericin-induced
graphs further revealed that the area occupied by the swollen
decrease of cathepsin activity was similar both in MN9D/neo
vacuoles in MN9D/f-Bak cells was significantly smaller than in
and MN9D/Bak cells suggesting that
MN9D/neo cells following nigericin treatment (Fig. 8C). Taken
Bak may maintain autophagic flux not by directly affecting ly-
together, we are tempted to argue that Bak may actively regu-
sosomal enzymatic activity but by other important steps of
late the vacuolar systems that are abnormally swollen during
autophagic events. Next, we monitored the ultrastructural
nigericin-induced cell death, and this activity may be involved
changes in MN9D/neo and MN9D/f-Bak cells after treatment
in maintaining autophagic flux during nigericin-induced cell
with or without nigericin. At 9 h, a slight difference in the num-
bers of AVs (white arrows) was detected in both MN9D/neoand MN9D/m-Bak cells. However, a large amount of swollen
vacuoles appeared in the cytosol of MN9D/neo cells (Fig. 8B,
Ultrastructural examination indicates that nigericin induces
black arrows). As indicated previously by Thorens and Roth
severe vacuolization in the cytosol, including autophagic vacu-
23278 JOURNAL OF BIOLOGICAL CHEMISTRY
VOLUME 287 • NUMBER 28 • JULY 6, 2012

Protective Role for Bak in Nigericin-induced Cell Death
Lamp1, we propose that lysosomal dysfunction may be respon-sible for nigericin-induced impairment of autophagic flux.
In this death paradigm, our data indicate that exogenous
expression of Bak reverses these phenomena. Consistent withour previously published data showing a dual role for Bax (29),for example, we demonstrate that Bak also renders a resistanceto nigericin-induced cell death and inhibits drug-induced accu-mulation of LC3-II. Moreover, the inhibitory role for Bak isfound in N18TG and HEK cells, indicating that its unusual pro-tective role is not cell type-specific. This inhibitory activity ofBak is carefully confirmed in various experimental conditions,including cells transiently or stably transfected with expressionvectors encoding one of several types of Bak or in Bak-silencedcells, indicating that Bak is indeed responsible for the inhibitionof nigericin-induced appearance of vacuoles, accumulation ofLC3-II, and subsequent cell death. Although accumulating evi-dence supports a protective role of autophagy in diverse condi-tions, immature completion of the autophagic process can also
lead to the impairment of autophagic flux, and this event is inturn linked to cell death (40). In this study, data from co-treat-ment with chloroquine and ultrastructural examination in Bak-overexpressing cells indicate that its inhibitory role may beclosely associated with its regulatory effect on maintaining
autophagic flux. Considering that maturation of lysosomesrequires the functional endosomal system (41, 42), Bak mayblock nigericin-induced generation of swollen vacuoles viainhibiting drug-induced impairment of the formation of late
endosome or lysosome compartments. However, this possibil-ity has to be thoroughly investigated.
There have been a few studies, including ones from our lab-
oratory, that have revealed that Bax and Bak delay or protectagainst cell death, depending on specific stimuli or develop-mental stages (29). Typically, Bax and Bak promote apoptosis
FIGURE 7. Knockdown of Bak accelerates nigericin-induced accumulation
through inducing mitochondrial outer membrane permeabili-
of LC3-II in HEK293 and MN9D cells. A and B, after 48 h of transient trans-
zation to release pro-apoptotic molecules from the mitochon-
fection with Myc-tagged human Bak (A) and negative (ctrl) siRNA or Bak siRNA
dria and causing excessive release of ER calcium (17, 43).
(B), HEK293 cells were treated with or without 1 M nigericin for 24 h. Immu-noblot analysis was then performed with anti-Myc, -Bak, -LC3, and -GAPDH
Indeed, we here demonstrate that transient or stable overex-
antibodies. Relative intensity of LC3-II in each condition was calculated and
pression of Bak accelerates staurosporine-induced MN9D cell
compared, as described above. C, after 24 h of transfection with empty vector
death, whereas it blocks nigericin-induced cell death. Etopo-
(pcDNA3.1) or a vector encoding FLAG-human Bak, MN9D cells were furthertransfected with negative siRNA or two distinct sets of siRNAs targeting
side-induced apoptotic cell death is also accelerated in Bak-
human Bak sequences (hBak). After 24 h post-transfection, MN9D cells were
overexpressing MN9D cells.3 This phenomenon is recapitu-
treated with 0.1 M nigericin for 18 h and subjected to immunoblot analysiswith anti-LC3, -FLAG, or -GAPDH antibodies. Relative intensity of LC3-II nor-
lated in other cell types, including N18TG cells, implying a dual
malized to GAPDH in each condition was calculated and compared. A–C, All
role for Bak in these distinct cell death paradigms. In consider-
values from quantitative analyses represent the mean ⫾ S.D. (error bars) from
ation of our data demonstrating that treatment of a pan-caspase
three independent experiments. *, p ⬍ 0.05; ***, p ⬍ 0.001.
inhibitor does not affect the nigericin-induced accumulation ofLC3-II and overexpressed Bak does not block nigericin-in-
oles and swollen vacuoles. Immunological analyses using anti-
duced levels of the cleaved caspase-3 in MN9D cells, it is highly
LC3 antibody show increase in LC3-II and a punctated staining
likely that the protective role for Bak in nigericin-induced cell
pattern in nigericin-treated cells, demonstrating that nigericin
death is independent of caspase activity. In contrast to what we
induces MN9D neurodegeneration, morphologically and bio-
expected, our unpublished data3 indicate that the protective
chemically typical of autophagy. Furthermore, our data for such
function of Bak during nigericin-induced cell death is also
lysomotropic agents as chloroquine and ammonium chloride
attributable to its BH3 domain, which has been demonstrated
support the notion that the nigericin-induced accumulation of
to be critical for exerting its pro-apoptotic activity. Although it
LC3-II is due to impaired autophgic flux. We found that treat-
has not been carefully determined whether deletion of the BH3
ment with another ionophore, monensin, that is structurally
domain also abolishes Bak-mediated inhibition of LC3-II accu-
and functionally related to nigericin, also impairs autophagicflux in MN9D cells. Based on our data from an in vitro cathepsinsubstrate assay and from immunocytochemical localization of
3 Y. Lee, J. Lim, I. J. Rhyu, and Y. J. Oh, unpublished data.
JULY 6, 2012 • VOLUME 287 • NUMBER 28
JOURNAL OF BIOLOGICAL CHEMISTRY

Protective Role for Bak in Nigericin-induced Cell Death
FIGURE 8. Bak attenuates nigericin-induced cell death by maintaining autophagic flux. A, MN9D cells stably overexpressing empty vector (MN9D/neo) or
FLAG-tagged Bak (MN9D/f-Bak) were treated with 0.1 M nigericin in the presence or the absence of 10 M CQ for the indicated time periods. Cell lysates wereseparated, transblotted, and probed with anti-LC3 and -GAPDH antibodies. Quantitation of the relative intensity of LC3-II normalized to GAPDH was carriedout. Each bar represents the mean ⫾ S.D. (error bars) from three independent experiments. B, ultrastructural photomicrographs of MN9D/neo and MN9D/f-Bakcells treated with or without 0.1 M nigericin for 9 h. Scale bar, 5 m. Autophagic vacuoles and swollen vacuoles in nigericin-treated cells were indicated bywhite arrows and by black arrows, respectively. Enlarged images of the same microscopic field in nigericin-treated MN9D/neo and MN9D/f-Bak cells are shownat the right. C, the area occupied by abnormally swollen vacuoles following nigericin treatment was measured with Scion Image. Values were shown as apercentage of the area occupied by swollen vacuoles over the total area of the cell. Thirty cells from two independent experiments were measured. *, p ⬍ 0.05;n.s., not significant.
mulation and swollen vacuole formation following nigericin
block drug-induced increased cytosolic Ca2⫹ level and conse-
treatment, it would be very intriguing to investigate how the
quently actively regulate autophagic flux. Because a common
BH3 domain distinctively plays a role in both apoptosis and
characteristic of neurodegenerative disorders is typically the
autophagy. The BH3 domain of Bak or Bax has been demon-
accumulation of protein aggregates, it would be very intriguing
strated to be required for interaction with Bcl-2 family proteins
to investigate whether Bak also plays a critical role in mainte-
or BH3-only proteins (17). Based on previous studies by others
nance of protein homeostasis in experimental models of
demonstrating that members of the Bcl-2 family actively influ-
Alzheimer, Parkinson, and Huntington disease. Through direct
ence autophagic events by regulating the rate of interaction
examination of how Bak is involved in regulating these events
with Beclin 1, another BH3-only protein (21–24, 44), it may be
and perhaps expansion of recent evidence of Bax regulatory
likely that mapping the interactions with Bcl-2 family members
activity on autophagy (46, 47), for example, we would be able to
or finding novel binding profiles of Bak may be the key to
gain a better understanding of the function of the Bcl-2 family
understanding the mechanisms by which Bak regulates nigeri-
proteins in the regulation of autophagy and of its potential as a
cin-induced autophagic cell death. The implication of Ca2⫹ in
novel therapeutic strategy in neurodegeneration.
autophagy has been also investigated (45). Our unpublisheddata3 indicated that the nigericin-induced increased level of
Acknowledgments—We thank Dr. Yoshimory (Osaka University) and
intracellular free Ca2⫹ is significantly attenuated in MN9D cells
Dr. Reed (Burnham Institute) for the generous gifts of GFP-tagged LC3
overexpressing Bak. Similarly, we observed that dysregulation
and human Bak cDNA, respectively. We thank Dr. Jürgen Roth (Uni-
of cytosolic Ca2⫹ can impair autophagic flux in MN9D cells.
versity of Zurich) for assistance in analyzing the electron micrographs
Under these conditions, therefore, we found that the chelation
in general and swollen vacuoles in particular.
of cytosolic Ca2⫹ or overexpression of calcium-binding proteinenhances autophagic flux during nigericin-induced neurode-
generation.3 Considering that a large amount of Bak is
1. Kroemer, G., Marin
˜o, G., and Levine, B. (2010) Autophagy and the inte-
expressed in the mitochondria of MN9D cells, we are tempted
grated stress response. Mol. Cell 40, 280 –293
to propose that the mitochondrially localized Bak seems to
2. He, C., and Klionsky, D. J. (2009) Regulation mechanisms and signaling
23280 JOURNAL OF BIOLOGICAL CHEMISTRY
VOLUME 287 • NUMBER 28 • JULY 6, 2012
Protective Role for Bak in Nigericin-induced Cell Death
pathways of autophagy. Annu. Rev. Genet. 43, 67–93
G., Herman, B., and Levine, B. (1998) Protection against fatal Sindbis virus
3. Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda,
encephalitis by Beclin, a novel Bcl-2-interacting protein. J. Virol. 72,
T., Kominami, E., Ohsumi, Y., and Yoshimori, T. (2000) LC3, a mamma-
lian homologue of yeast Apg8p, is localized in autophagosome mem-
20. Maiuri, M. C., Le Toumelin, G., Criollo, A., Rain, J. C., Gautier, F., Juin, P.,
branes after processing. EMBO J. 19, 5720 –5728
Tasdemir, E., Pierron, G., Troulinaki, K., Tavernarakis, N., Hickman, J. A.,
4. Tanida, I., Minematsu-Ikeguchi, N., Ueno, T., and Kominami, E. (2005)
Geneste, O., and Kroemer, G. (2007) Functional and physical interaction
Lysosomal turnover, but not a cellular level, of endogenous LC3 is a
and a BH3-like domain in Beclin-1. EMBO J. 26,
marker for autophagy. Autophagy 1, 84 –91
5. Eskelinen, E. L. (2005) Maturation of autophagic vacuoles in mammalian
21. Høyer-Hansen, M., Bastholm, L., Szyniarowski, P., Campanella, M., Sz-
cells. Autophagy 1, 1–10
abadkai, G., Farkas, T., Bianchi, K., Fehrenbacher, N., Elling, F., Rizzuto, R.,
6. Fader, C. M., and Colombo, M. I. (2009) Autophagy and multivesicular
Mathiasen, I. S., and Jäättelä, M. (2007) Control of macroautophagy by
bodies. Two closely related partners. Cell Death Differ. 16, 70 –78
calcium, calmodulin-dependent kinase kinase-, and Bcl-2. Mol. Cell 25,
7. Filimonenko, M., Stuffers, S., Raiborg, C., Yamamoto, A., Malerød, L.,
Fisher, E. M., Isaacs, A., Brech, A., Stenmark, H., and Simonsen, A. (2007)
22. Strappazzon, F., Vietri-Rudan, M., Campello, S., Nazio, F., Florenzano, F.,
Functional multivesicular bodies are required for autophagic clearance of
Fimia, G. M., Piacentini, M., Levine, B., and Cecconi, F. (2011) Mitochon-
protein aggregates associated with neurodegenerative disease. J. Cell Biol.
drial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 30,
8. Jäger, S., Bucci, C., Tanida, I., Ueno, T., Kominami, E., Saftig, P., and
23. Erlich, S., Mizrachy, L., Segev, O., Lindenboim, L., Zmira, O., Adi-Harel,
Eskelinen, E. L. (2004) Role for Rab7 in maturation of late autophagic
S., Hirsch, J. A., Stein, R., and Pinkas-Kramarski, R. (2007) Differential
vacuoles. J. Cell Sci. 117, 4837– 4848
interactions between Beclin 1 and Bcl-2 family members. Autophagy 3,
9. Lee, J. A., Beigneux, A., Ahmad, S. T., Young, S. G., and Gao, F. B. (2007)
ESCRT-III dysfunction causes autophagosome accumulation and neuro-
24. Malik, S. A., Orhon, I., Morselli, E., Criollo, A., Shen, S., Marino, G., Be-
degeneration. Curr. Biol. 17, 1561–1567
nyounes, A., Benit, P., Rustin, P., Maiuri, M. C., and Kroemer, G. (2011)
10. Galluzzi, L., Aaronson, S. A., Abrams, J., Alnemri, E. S., Andrews, D. W.,
BH3 mimetics activate multiple pro-autophagic pathways. Oncogene 30,
Baehrecke, E. H., Bazan, N. G., Blagosklonny, M. V., Blomgren, K., Borner,
C., Bredesen, D. E., Brenner, C., Castedo, M., Cidlowski, J. A., Ciecha-
25. Steinrauf, L. K., Czerwinski, E. W., and Pinkerton, M. (1971) Comparison
nover, A., Cohen, G. M., De Laurenzi, V., De Maria, R., Deshmukh, M.,
of the monovalent cation complexes of monensin, nigericin, and diane-
Dynlacht, B. D., El-Deiry, W. S., Flavell, R. A., Fulda, S., Garrido, C., Gol-
mycin. Biochem. Biophys. Res. Commun. 45, 1279 –1283
stein, P., Gougeon, M. L., Green, D. R., Gronemeyer, H., Hajno´czky, G.,
26. Zhang, L., Yu, J., Pan, H., Hu, P., Hao, Y., Cai, W., Zhu, H., Yu, A. D., Xie,
Hardwick, J. M., Hengartner, M. O., Ichijo, H., Jäättelä, M., Kepp, O.,
X., Ma, D., and Yuan, J. (2007) Small molecule regulators of autophagy
Kimchi, A., Klionsky, D. J., Knight, R. A., Kornbluth, S., Kumar, S., Levine,
identified by an image-based high-throughput screen. Proc. Natl. Acad.
B., Lipton, S. A., Lugli, E., Madeo, F., Malomi, W., Marine, J. C., Martin,
Sci. U.S.A. 104, 19023–19028
S. J., Medema, J. P., Mehlen, P., Melino, G., Moll, U. M., Morselli, E.,
27. Grinde, B. (1983) Effect of carboxylic ionophores on lysosomal protein
Nagata, S., Nicholson, D. W., Nicotera, P., Nunez, G., Oren, M., Pen-
degradation in rat hepatocytes. Exp. Cell Res. 149, 27–35
ninger, J., Pervaiz, S., Peter, M. E., Piacentini, M., Prehn, J. H., Puthalakath,
28. Kawai, A., Uchiyama, H., Takano, S., Nakamura, N., and Ohkuma, S.
H., Rabinovich, G. A., Rizzuto, R., Rodrigues, C. M., Rubinsztein, D. C.,
(2007) Autophagosome-lysosome fusion depends on the pH in acidic
Rudel, T., Scorrano, L., Simon, H. U., Steller, H., Tschopp, J., Tsujimoto,
compartments in CHO cells. Autophagy 3, 154 –157
Y., Vandenabeele, P., Vitale, I., Vousden, K. H., Youle, R. J., Yuan, J., Zhi-
29. Oh, J. H., O'Malley, K. L., Krajewski, S., Reed, J. C., and Oh, Y. J. (1997) Bax
votovsky, B., and Kroemer, G. (2009) Guidelines for the use and interpre-
accelerates staurosporine-induced but suppresses nigericin-induced neu-
tation of assays for monitoring cell death in higher eukaryotes. Cell Death
ronal cell death. Neuroreport 8, 1851–1856
Differ. 16, 1093–1107
30. Kim, J. E., Oh, J. H., Choi, W. S., Chang, I. I., Sohn, S., Krajewski, S., Reed,
11. Scarlatti, F., Granata, R., Meijer, A. J., and Codogno, P. (2009) Does au-
J. C., O'Malley, K. L., and Oh, Y. J. (1999) Sequential cleavage of poly(ADP-
tophagy have a license to kill mammalian cells? Cell Death Differ. 16,
ribose)polymerase and appearance of a small Bax-immunoreactive pro-
tein are blocked by Bcl-X and caspase inhibitors during staurosporine-
12. Kroemer, G., and Levine, B. (2008) Autophagic cell death. The story of a
induced dopaminergic neuronal apoptosis. J. Neurochem. 72, 2456 –2463
misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004 –1010
31. Lee, Y. M., Park, S. H., Shin, D. I., Hwang, J. Y., Park, B., Park, Y. J., Lee,
13. Eisenberg-Lerner, A., Bialik, S., Simon, H. U., and Kimchi, A. (2009) Life
T. H., Chae, H. Z., Jin, B. K., Oh, T. H., and Oh, Y. J. (2008) Oxidative
and death partners. Apoptosis, autophagy, and the cross-talk between
modification of peroxiredoxin is associated with drug-induced apoptotic
them. Cell Death Differ. 16, 966 –975
signaling in experimental models of Parkinson disease. J. Biol. Chem. 283,
14. Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S.,
Baehrecke, E. H., Blagosklonny, M. V., El-Deiry, W. S., Golstein, P., Green,
32. Lim, J., Kim, H. W., Youdim, M. B., Rhyu, I. J., Choe, K. M., and Oh, Y. J.
D. R., Hengartner, M., Knight, R. A., Kumar, S., Lipton, S. A., Malorni, W.,
(2011) Binding preference of p62 toward LC3-II during dopaminergic
˜ez, G., Peter, M. E., Tschopp, J., Yuan, J., Piacentini, M., Zhivotovsky,
neurotoxin-induced impairment of autophagic flux. Autophagy 7, 51– 60
B., Melino, G., and the Nomenclature Committee on Cell Death 2009
33. Hansen, M. B., Nielsen, S. E., and Berg, K. (1989) Re-examination and
(2009) Classification of cell death. Recommendations of the Nomencla-
further development of a precise and rapid dye method for measuring cell
ture Committee on Cell Death 2009. Cell Death Differ. 16, 3–11
growth/cell kill. J. Immunol. Methods 119, 203–210
15. Maiuri, M. C., Zalckvar, E., Kimchi, A., and Kroemer, G. (2007) Self-eating
34. Ylä-Anttila, P., Vihinen, H., Jokitalo, E., and Eskelinen, E. L. (2009) Mon-
and self-killing. Cross-talk between autophagy and apoptosis. Nat. Rev.
itoring autophagy by electron microscopy in mammalian cells. Methods
Mol. Cell Biol. 8, 741–752
Enzymol. 452, 143–164
16. Gross, A., McDonnell, J. M., and Korsmeyer, S. J. (1999) BCL-2 family
35. Thorens, B., and Roth, J. (1996) Intracellular targeting of GLUT4 in trans-
members and the mitochondria in apoptosis. Genes Dev. 13, 1899 –1911
fected insulinoma cells. Evidence for association with constitutively recy-
17. Chipuk, J. E., Moldoveanu, T., Llambi, F., Parsons, M. J., and Green, D. R.
cling vesicles distinct from synaptophysin and insulin vesicles. J. Cell Sci.
(2010) The BCL-2 family reunion. Mol. Cell 37, 299 –310
18. Kim, H., Tu, H. C., Ren, D., Takeuchi, O., Jeffers, J. R., Zambetti, G. P.,
36. Iwai-Kanai, E., Yuan, H., Huang, C., Sayen, M. R., Perry-Garza, C. N., Kim,
Hsieh, J. J., and Cheng, E. H. (2009) Stepwise activation of BAX and BAK
L., and Gottlieb, R. A. (2008) A method to measure cardiac autophagic flux
by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 36,
in vivo. Autophagy 4, 322–329
37. Komatsu, M., Waguri, S., Koike, M., Sou, Y. S., Ueno, T., Hara, T., Miz-
19. Liang, X. H., Kleeman, L. K., Jiang, H. H., Gordon, G., Goldman, J. E., Berry,
ushima, N., Iwata, J., Ezaki, J., Murata, S., Hamazaki, J., Nishito, Y., Iemura,
JULY 6, 2012 • VOLUME 287 • NUMBER 28
JOURNAL OF BIOLOGICAL CHEMISTRY
Protective Role for Bak in Nigericin-induced Cell Death
S., Natsume, T., Yanagawa, T., Uwayama, J., Warabi, E., Yoshida, H., Ishii,
Mol. Membr. Biol. 20, 141–154
T., Kobayashi, A., Yamamoto, M., Yue, Z., Uchiyama, Y., Kominami, E.,
43. Tait, S. W., and Green, D. R. (2010) Mitochondria and cell death. Outer
and Tanaka, K. (2007) Homeostatic levels of p62 control cytoplasmic in-
membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11,
clusion body formation in autophagy-deficient mice. Cell 131, 1149 –1163
38. Pinkerton, M., and Steinrauf, L. K. (1970) Molecular structure of monova-
44. Maiuri, M. C., Criollo, A., Tasdemir, E., Vicencio, J. M., Tajeddine, N.,
lent metal cation complexes of monensin. J. Mol. Biol. 49, 533–546
Hickman, J. A., Geneste, O., and Kroemer, G. (2007) BH3-only proteins
39. Boya, P., Gonza´lez-Polo, R. A., Casares, N., Perfettini, J. L., Dessen, P.,
and BH3 mimetics induce autophagy by competitively disrupting the in-
Larochette, N., Me´tivier, D., Meley, D., Souquere, S., Yoshimori, T., Pier-
teraction between Beclin 1 and Bcl-2/Bcl-X . Autophagy 3, 374 –376
ron, G., Codogno, P., and Kroemer, G. (2005) Inhibition of macroau-
45. Høyer-Hansen, M., and Jäättelä, M. (2007) Connecting endoplasmic re-
tophagy triggers apoptosis. Mol. Cell Biol. 25, 1025–1040
ticulum stress to autophagy by unfolded protein response and calcium.
40. Wong, E., and Cuervo, A. M. (2010) Autophagy gone awry in neurodegen-
Cell Death Differ. 14, 1576 –1582
erative diseases. Nat. Neurosci. 13, 805– 811
46. Yee, K. S., Wilkinson, S., James, J., Ryan, K. M., and Vousden, K. H. (2009)
41. Cook, N. R., Row, P. E., and Davidson, H. W. (2004) Lysosome-associated
PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death
membrane protein 1 (Lamp1) traffics directly from the TGN to early en-
Differ. 16, 1135–1145
dosomes. Traffic 5, 685– 699
47. Luo, S., and Rubinsztein, D. C. (2010) Apoptosis blocks Beclin 1-depen-
42. Luzio, J. P., Poupon, V., Lindsay, M. R., Mullock, B. M., Piper, R. C., and
dent autophagosome synthesis. An effect rescued by Bcl-xL. Cell Death
Pryor, P. R. (2003) Membrane dynamics and the biogenesis of lysosomes.
Differ. 17, 268 –277
23282 JOURNAL OF BIOLOGICAL CHEMISTRY
VOLUME 287 • NUMBER 28 • JULY 6, 2012
Source: http://web.yonsei.ac.kr/neurolab/published/70.pdf
RA Journal of Applied Research Volume 1 Issue 1 Pages-18-43 November -2014 ISSN (e): Appli Importance of Physicochemical Properties In Drug Discovery. (Review Article) Kapadia Akshay Bhupendra Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology, Nathalal Parekh Marg, Matunga (East), Mumbai.
DANUBE SYMPOSIUM OF NEPHROLOGY and ERA-EDTA CME-Literature-Update-Course Krems - AustriaAugust 28. – 30. 2008 XIXth Danube Symposium of Nephrology Dear colleagues, dear friends, dear participants of the XIXth Danube Symposium of Nephrology in Krems, On behalf of the Danube Society of Nephrology (DSN), it is my great pleasure and It is a great pleasure and honour for me to welcome you all here in Krems – a very nice